


We thank Finsterer et al. for their pertinent comments on our report of the diagnosis of left ventricular noncompaction (LVNC) in a young adult female with Fabry disease,1 and for the opportunity to describe in more detail her clinical history, which necessarily had to be concise in the original case report format, and to elaborate on the diagnostic value and interpretation of endomyocardial biopsy in females with Fabry disease.
This patient belongs to a large three-generation family with classical Fabry disease (Figure 1) segregating with a relatively common nonsense mutation in the alpha-galactosidase gene (GLA) that leads to the premature termination of GLA mRNA translation at codon 220 (p.R220X).2 Because it severely compromises residual alpha-galactosidase enzyme activity, the p.R220X mutation is associated with the classical phenotype of Fabry disease.3 The GLA gene locus is on the X chromosome, and therefore the inheritance of Fabry disease follows an X-linked pattern with more severe and earlier clinical manifestations in hemizygous males, and more variable and organ-restricted phenotypes in heterozygous females.2 This is well exemplified by our patient's family, where the affected males in the second generation presented with the classical phenotype of Fabry disease and developed end-stage kidney disease long before the availability of enzyme replacement therapy (ERT), while her affected sisters were asymptomatic or manifested partial clinical phenotypes at later ages.
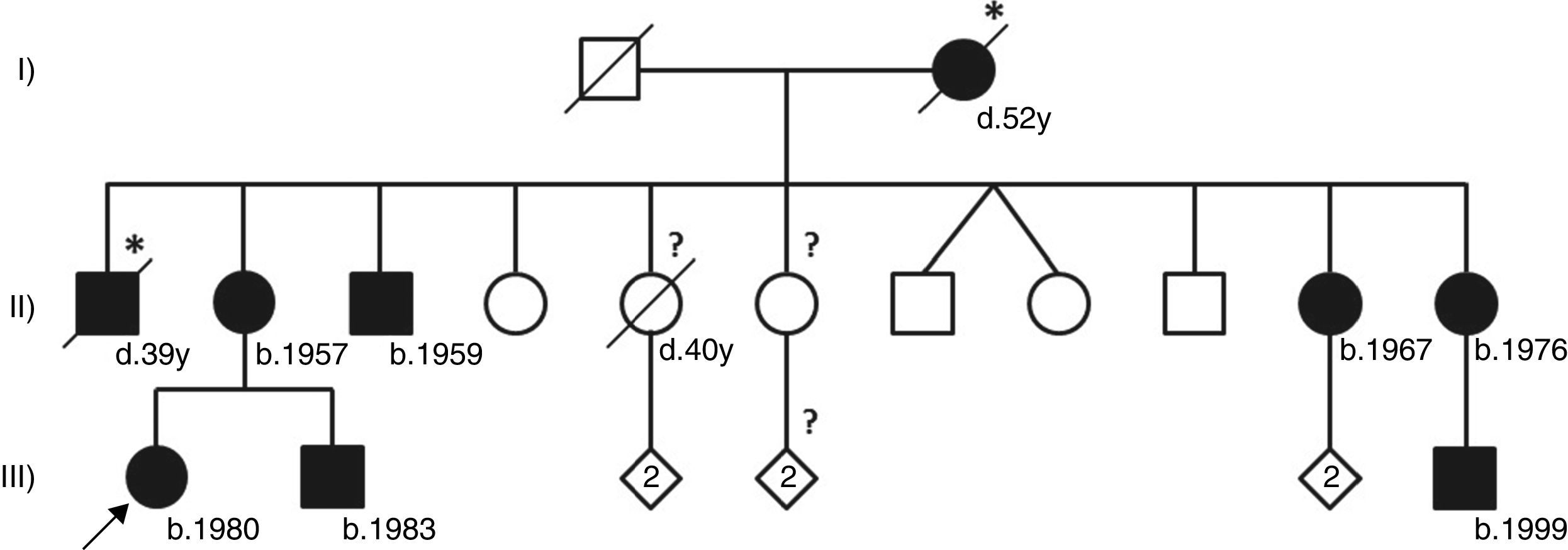
Pedigree of the reported Fabry disease patient and of her family. Squares indicate males, circles indicate females, solid black symbols indicate individuals diagnosed with Fabry disease; symbols with a diagonal line denote deceased individuals. Asterisks indicate a clinical or pedigree-based diagnosis of Fabry disease. Diamonds indicate healthy sibs of different genders, with their number specified by the cardinal inside the symbol. The arrow indicates the patient presented in our paper. Question-marks indicate individuals who have not been clinically or genetically assessed for Fabry disease. The two diagonal vertical lines originating from the same point indicate dizygotic twins. Year of birth is indicated by “b.”; age at death, in full years (“y”), is indicated by “d.”. Patient I-2 was reported to have died from “kidney disease”; patient II-1 was reported to have died from “cerebrovascular disease”, nine years after receiving a kidney transplant; patient II-5 had type 2 diabetes mellitus and died from acute stroke. All the affected males presented with the classical phenotype of Fabry disease. Patient II-3, who is followed at another hospital, started hemodialysis at age 35 and received a kidney transplant about two years later. Patients II-11 and III-9 live abroad and are also followed elsewhere. Patients II-2, II-10 and III-2, who are under our clinical care, started enzyme replacement therapy in 2005, 2002 and 2003, respectively. Neither of the two younger sisters of the proband's mother has yet presented any major cardiac or cerebrovascular complications of Fabry disease. Individuals II-4, II-7, II-8, II-9, III-3/4 and III-7/8 have been genetically screened and did not carry the GLA p.R220X mutation.
Both the mother of our patient and one of her younger sisters were diagnosed with Fabry nephropathy on kidney biopsies obtained at ages 45 and 32 years, respectively,4 but neither of them manifested any other complications of Fabry disease at the baseline evaluation. The GLA mutation p.R220X was originally identified in a 56-year-old Australian woman presenting with hypertrophic cardiomyopathy5 and typical histopathological findings of Fabry disease were observed in renal and myocardial necropsy specimens from a 71-year-old Japanese woman heterozygous for the same mutation.6 On the whole, these cases illustrate the clinical variability of Fabry disease in affected females, even when they carry highly pathogenic GLA mutations. As in other X-linked recessive Mendelian disorders, the most striking example of the phenotypic variability of Fabry disease in females is the discordant expression in monozygotic twins,7 due to highly unbalanced X-chromosome inactivation in opposite directions, with the parental X chromosome carrying the pathogenic GLA mutation preferentially active in the clinically affected twin and the X chromosome inherited from the other parent preferentially active in the asymptomatic twin sister.
However, proper interpretation of the effect of X-chromosome inactivation (lyonization) on the clinical phenotypes seen in females carrying X-linked mutant genes8 should take into consideration not only the overall ratio of maternal-to-paternal X-chromosome inactivation which occurs at a very early stage in embryonic development, but also the subsequent random cell assortment during organogenesis9 that may lead to discordant proportions of cells with one or the other functional X chromosome in different tissues. In solid organs, like the heart, the outcome of this process at the cellular level is the mosaic expression of X-linked phenotypes, topographically correlated with X-inactivation patches,10 i.e., clonally related contiguous groups of cells with the same functionally inactivated parental X chromosome. We are not aware of any published studies on cardiomyocyte X-inactivation patches in the human heart, but in the mouse they can be large.11
Bearing in mind the above considerations about lyonization, the endomyocardial biopsy unequivocally demonstrated that our patient has Fabry cardiomyopathy, having at least one patch of affected cardiomyocytes in the apical region of the right side of the interventricular septum, from where the biopsy was taken (but a normal biopsy result would not have excluded the diagnosis!); it is quite possible that the sampled patch of affected cardiomyocytes extends to the hypertrabeculated apex of the left ventricle (LV). A likely explanation for the discordant histopathological expression of Fabry disease in cardiomyocytes and endothelial cells is the different progenitor cell populations from which they arise.12
Regarding the argument raised by Finsterer et al. that our patient may have a second heart disorder, pathogenically unrelated to Fabry disease, the following information is important: (i) all the affected individuals that were alive in this family underwent comprehensive, multidisciplinary assessment at baseline, and have been followed according to expert guidelines for the recognition, evaluation, and surveillance of disease-associated morbidities13; (ii) our patient has no clinical manifestations whatsoever of a coexisting neuropathic or myopathic disorder; (iii) her first routine echocardiogram, which had been performed at age 29 years, was unremarkable, but the acoustic window was poor, preventing the exclusion of LV hypertrabeculation at that time; (iv) the patient's mother had no signs of LVNC on the echocardiogram performed at the age of 48 years, for baseline evaluation before initiating ERT, and presented only mild LV hypertrophy at her most recent echocardiographic assessment, at age 56 years; (v) the 72-year-old father of the patient, who is followed at another hospital for severe coronary heart disease, past history of myocardial infarction and LV dysfunction, also has no echocardiographic signs of LVNC or hypertrabeculation. We entirely agree with Finsterer et al. concerning the clinical importance of detailed neuromuscular assessment for the differential diagnosis of cardiomyopathies,14 including acquired LVNC,15 and our patient has been evaluated according to such diagnostic concerns.
LVNC is a rare condition that can occur in isolation or coexist with other cardiac and/or systemic anomalies.16,17 The majority of cases are congenital and are thought to result from an arrest of the normal compaction process of the myocardium during fetal development. The familial types of LVNC are the most common and follow autosomal-dominant, X-linked, or mitochondrial-inheritance patterns.16 However, since our patient is a sporadic case of apparently acquired LV hypertrabeculation, the diagnosis of a familial type of LVNC is quite unlikely.
As acknowledged by Finsterer et al., LV hypertrabeculation has been reported in patients with other lysosomal storage disorders (LSDs), including Pompe disease14 and Danon disease.18 We suggest that Fabry disease should be added to the list of LSDs that can give rise to acquired LVNC. Prenatal hypertrabeculation and noncompaction of the ventricular wall were observed in a mouse model of severe glycogen storage disease type IV (Andersen disease),3 in association with increased expression of cell-cycle regulators in cardiomyocytes. Whether similar molecular mechanisms are also operative in LSD-related acquired LVNC is as yet unknown. As recently addressed in a state-of-art paper,17 further research is needed to clarify whether LVNC is a primary genetic cardiomyopathy or a morphologic trait shared by different cardiomyopathies, as well as to elucidate their molecular pathogenesis.
Conflicts of interestThe authors have no conflicts of interest to declare.
