



Hypercholesterolemia results from an alteration, genetic or acquired, in lipoprotein metabolism. Evidence that hypercholesterolemia is associated with the atherosclerotic process from childhood justifies the screening of high-risk children and initiation of therapy at preschool ages.
ObjectiveTo assess children referred for pediatric consultations due to hypercholesterolemia.
MethodsChildren and adolescents referred for pediatric consultations with a diagnosis of hypercholesterolemia were enrolled. Information on family history and clinical, anthropometric and biochemical parameters was recorded and, when appropriate, molecular study was performed.
ResultsA total of 168 children were assessed. Forty-six presented a familial hypercholesterolemia phenotype and in 22 of these, a mutation in the low-density lipoprotein (LDL) receptor gene was identified. The lipid profile of the group with mutations showed significantly higher values of total and non-high-density lipoprotein (HDL) cholesterol compared to the group without mutations (total cholesterol 316.5±75.9 mg/dl vs. 260.9±42.0 mg/dl; non-HDL cholesterol 268.3±72.6 mg/dl vs. 203.5±43.9 mg/dl; p<0.05). Of the total, 55 were prescribed pharmacological therapy and the others underwent diet and exercise interventions only. A greater reduction in LDL cholesterol was observed in individuals under pharmacological therapy compared to those prescribed diet and exercise only (30.3% vs. 18.1%). Drug side effects were insignificant.
ConclusionIt is possible to maintain a normal lipid profile in most individuals with familial hypercholesterolemia in order to reduce the risk of early onset of atherosclerosis, which is associated with serious cardiovascular complications from childhood.
A hipercolesterolemia resulta de uma alteração do metabolismo das lipoproteínas e pode ter uma origem ambiental ou genética, como é o caso da hipercolesterolemia familiar. A evidência de que a hipercolesterolemia se associa ao processo aterosclerótico desde a idade pediátrica justifica o rastreio obrigatório em todas as crianças de risco e a instituição de terapêutica individualizada desde a idade pré-escolar.
ObjetivoAvaliar as crianças referenciadas por hipercolesterolemia à consulta de Pediatria.
MétodoForam incluídas as crianças e adolescentes referenciados à consulta de Pediatria por hipercolesterolemia. Procedeu-se à avaliação da história familiar e de parâmetros antropométricos, clínicos e bioquímicos e, quando justificado, ao estudo molecular.
ResultadosForam avaliadas 168 crianças. Apresentavam fenótipo de hipercolesterolemia familiar 46 crianças e, em 22 destas, foi identificada uma mutação no gene do recetor das lipoproteínas de baixa densidade. O perfil lipídico revelou valores significativamente superiores de colesterol total e colesterol n-HDL no grupo com mutação relativamente ao sem mutação (colesterol total 316,5 ± 75,9 mg/dL versus 260,9 ± 42,0 mg/dL; colesterol n-HDL 268,3 ± 72,6 mg/dL versus 203,5 ± 43,9 mg/dL; p < 0,05). Do total das crianças, 55 efetuaram terapêutica farmacológica, as restantes apenas intervenção comportamental. Registou-se maior redução do c-LDL nos indivíduos com intervenção farmacológica relativamente aos sujeitos apenas a alterações comportamentais (30,3 versus 18,1%). Os efeitos laterais dos fármacos foram desprezíveis.
ConclusãoÉ possível manter um perfil lipídico sustentado em valores normais na maioria dos indivíduos com hipercolesterolemia familiar, de forma a reduzir o risco de evolução precoce do processo aterosclerótico, comprovadamente associado a graves complicações cardiovasculares logo desde a idade pediátrica.
Cardiovascular disease is the leading cause of mortality and morbidity in developed countries.1,2
Studies such as PDAY (Pathobiological Determinants of Atherosclerosis in Youth) and the Bogalusa Heart Study have shown that the atherosclerotic process begins in childhood, and that both genetic and environmental factors, particularly diet and exercise, play a role.2–6
The principal risk factors for cardiovascular disease in adulthood are high low-density lipoprotein cholesterol (LDL-C), low high-density lipoprotein cholesterol (HDL-C), hypertension, type 1 and 2 diabetes, smoking and obesity.1–3 Many of these factors are manifested in childhood, which justifies intervention at early ages for cardiovascular disease prevention.1–3
Hypercholesterolemia results from alterations in lipoprotein metabolism that lead to high total cholesterol, LDL-C or triglycerides, and/or low HDL-C. Most children with dyslipidemia have an idiopathic form (polygenic, risk factor-associated or multifactorial), whereas a minority will have monogenic forms, such as familial hypercholesterolemia (FH) or secondary dyslipidemias.
FH is an autosomal dominant disease, with a prevalence of 1/500 in European populations, that results from a mutation in one of three genes: LDLR, APOB and PCSK9. It is characterized by high plasma levels of LDL-C, which is deposited in arteries, leading to premature development of atherosclerosis and cardiovascular disease.8 Serum lipid and lipoprotein levels increase progressively during infancy, approaching adult levels by the age of two. Levels subsequently decrease during puberty, only to rise again to adult values.2,7 This has implications for hypercholesterolemia screening, and it is recommended that high-risk children be screened after the age of two.2
Evidence that hypercholesterolemia is associated with the atherosclerotic process from childhood and is accompanied by increased prevalence of cardiovascular risk factors, particularly obesity and metabolic syndrome, justifies screening for dyslipidemia in children and initiation of therapy at preschool ages.3,9
The objective of this study was to assess the clinical and laboratory parameters of children referred for pediatric consultations due to hypercholesterolemia.
MethodsChildren and adolescents referred for outpatient consultations in the pediatric nutrition unit at a tertiary hospital due to suspected hypercholesterolemia were included in the study.
Information was recorded on family history (cardiovascular disease, serum total cholesterol, LDL-C and triglycerides), anthropometric assessment at all consultations (weight, height, body mass index [BMI] and percentage fat mass), lipid profile after a minimum of 12 hours fasting (total cholesterol, LDL-C, HDL-C, triglycerides, apolipoproteins A1 and B [Apo A1 and Apo B] and lipoprotein(a) [Lp(a)]). Children with FH phenotype underwent molecular study. The treatment prescribed was analyzed, namely behavior modification and pharmacological therapy (drugs used, efficacy and side effects, the latter being evaluated as recommended).
Percentage fat mass was determined by bioimpedance in all patients aged six or over.
Cases suggestive of FH were selected on the basis of criteria adapted from the Simon Broome Heart Research Trust (Table 1).10
Criteria for familial hypercholesterolemia, adapted from those of the Simon Broome Heart Research Trust.
| Confirmed familial hypercholesterolemia: |
| • Index case: child under 16 with total cholesterol >260 mg/dl (6.7 mmol/l) or LDL cholesterol >155 mg/dl (4.0 mmol/l);• Index case: adult with total cholesterol >290 mg/dl (7.5 mmol/l) or LDL cholesterol >190 mg/dl (4.9 mmol/l),and• Tendon xanthoma in the index case or relative (parents, children, grandparents, siblings, aunts or uncles)or• Genetic evidence of a mutation in LDLR or APOB gene |
| Possible familial hypercholesterolemia: |
| • Index case: child under 16 with total cholesterol >260 mg/dl (6.7 mmol/l) or LDL cholesterol >155 mg/dl (4.0 mmol/l);• Index case: adult with total cholesterol >290 mg/dl (7.5 mmol/l) or LDL cholesterol >190 mg/dl (4.9 mmol/l),and• Family history of myocardial infarction before the age of 50 in grandparents or aunts or uncles, or before the age of 60 in parents, siblings or children, and/or family history of elevated cholesterol (>290 mg/dl) in parents, siblings or children, or total cholesterol >290 mg/dl (7.5 mmol/l) in grandparents and/or aunts or uncles. |
APOB: apolipoprotein B; LDL: low-density lipoprotein; LDLR: low-density lipoprotein receptor.
These cases were referred to the Portuguese Familial Hypercholesterolemia Study for molecular study of the index case, parents and other relatives. Genetic study included screening for mutations in the genes coding for LDL receptors (LDLR) and Apo B (APOB), and large rearrangements in the LDLR gene; if no mutation was identified in these genes, patients were screened for point mutations in the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene.11–14 The mere presence of a mutation in the LDLR gene was not taken to imply that it is the cause of the disease. Familial studies were performed to confirm cosegregation of the identified mutation with hypercholesterolemia in order to demonstrate the pathogenicity of the alteration. The frequency of alleles in the apolipoprotein E gene (APOE) was also determined.
The data were analyzed using SPSS version 19.0. The Student's t test was used for comparisons and the Kruskal–Wallis test for nonparametric variables.
ResultsA total of 168 children and adolescents were assessed, equally distributed between the sexes (females 51.1%). Mean age at first consultation was 8.7 years (median 9.0 years), and minimum age at the time of referral was 2.5 years.
Anthropometric evaluation of the total population at first consultation revealed a mean BMI z-score of 1.6 ± 1.0.
Assessment of lipid profile showed mean total cholesterol of 242.2±60.1 mg/dl (124.0–552.0 mg/dl) and mean non-HDL cholesterol of 191.8±60.3 mg/dl (94.0–472.3 mg/dl).
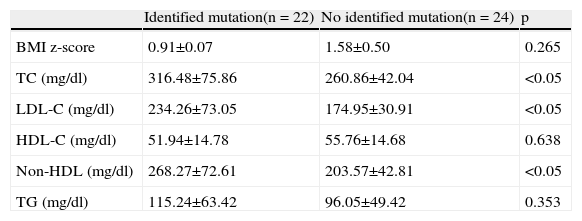
Forty-six children presented an FH phenotype, and in 22 of these a mutation in the LDLR gene was identified. No mutations were identified in the APOB or PCSK9 genes. Screening for APOE polymorphisms revealed the following allele frequencies: ¿2 3.7%, ¿3 78.0%, and ¿4 18.3%. Allele ¿4 was less frequent in individuals with an LDLR mutation (23.9% vs. 45.0%) (p=0.153).
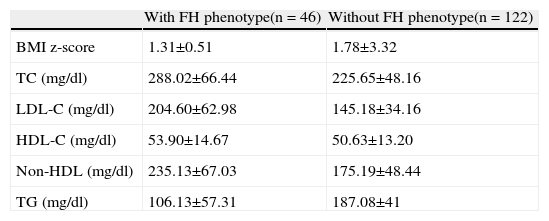
Comparison of nutritional status at the first consultation of patients with the FH phenotype revealed no significant differences between those with and without a mutation (mean BMI z-score 0.73±1.56 vs. 1.43±2.34, p=0.265) (Tables 2 and 3).
Body mass index and lipid profile (means ± standard deviation) in patients with and without familial hypercholesterolemia phenotype.
| With FH phenotype(n = 46) | Without FH phenotype(n = 122) | |
| BMI z-score | 1.31±0.51 | 1.78±3.32 |
| TC (mg/dl) | 288.02±66.44 | 225.65±48.16 |
| LDL-C (mg/dl) | 204.60±62.98 | 145.18±34.16 |
| HDL-C (mg/dl) | 53.90±14.67 | 50.63±13.20 |
| Non-HDL (mg/dl) | 235.13±67.03 | 175.19±48.44 |
| TG (mg/dl) | 106.13±57.31 | 187.08±41 |
BMI: body mass index; HDL-C: high density lipoprotein cholesterol; LDL-C: low density lipoprotein cholesterol; Non-HDL: total cholesterol except high density lipoprotein; TC: total cholesterol; TG: triglycerides.
Body mass index and lipid profile (means ± standard deviation) in patients with familial hypercholesterolemia phenotype, with and without an identified mutation.
| Identified mutation(n = 22) | No identified mutation(n = 24) | p | |
| BMI z-score | 0.91±0.07 | 1.58±0.50 | 0.265 |
| TC (mg/dl) | 316.48±75.86 | 260.86±42.04 | <0.05 |
| LDL-C (mg/dl) | 234.26±73.05 | 174.95±30.91 | <0.05 |
| HDL-C (mg/dl) | 51.94±14.78 | 55.76±14.68 | 0.638 |
| Non-HDL (mg/dl) | 268.27±72.61 | 203.57±42.81 | <0.05 |
| TG (mg/dl) | 115.24±63.42 | 96.05±49.42 | 0.353 |
BMI: body mass index; HDL-C: high density lipoprotein cholesterol; LDL-C: low density lipoprotein cholesterol; Non-HDL: total cholesterol except high density lipoprotein; TC: total cholesterol; TG: triglycerides.
Lipid profile showed significantly higher values in the group with a mutation compared to those without but with FH phenotype (total cholesterol 316.5±75.9 mg/dl vs. 260.9±42.0 mg/dl, and non-HDL cholesterol 268.3±72.6 mg/dl vs. 203.5±43.9 mg/dl; p<0.05) (Table 3, Figures 1 and 2).
.")
Of the total of 168 children, 113 were prescribed behavior modification only, including dietary measures designed to reduce total and saturated fat and cholesterol intake and efforts to increase exercise levels. The other 55 patients were prescribed pharmacological therapy as well as behavior modification.
Pharmacological therapy was required in 73.9% of patients with an FH phenotype, as opposed to 16.4% of those without. Of those with FH phenotype, pharmacological therapy was needed in 90.9% of those with a mutation and in 58.3% of those with no identified mutation.
There was a stronger response to behavior modification in individuals with APOE allele ¿4, of whom 57.1% required pharmacological therapy, as opposed to 74.1% of those without (p<0.05).
The reduction in LDL-C was greater in those under pharmacological therapy compared to those with behavior modification only (31.1% vs. 18.7%; p<0.05) (Figure 3).
.")
Drug therapy included bile acid binding resins in 37 patients, statins in 23 and ezetimibe in 20; combined therapy with statins and ezetimibe was prescribed in 13. The statin used in this patient population was pravastatin.
Use of resins was accompanied by gastrointestinal symptoms such as constipation and flatulence, together with poor compliance due to their taste and granular consistency. Side effects prompted discontinuation of statin therapy in one adolescent due to elevation of muscle enzymes (peak creatine kinase of 3588 U/l), although the patient remained asymptomatic.
Anthropometric assessment of the total population at the first consultation showed a mean BMI z-score of 1.6±1.0. Comparison of nutritional status at first consultation of patients with FH phenotype revealed no significant differences between those with and without a mutation (mean BMI z-score 0.73±1.56 vs. 1.43±2.34, p<0.265).
DiscussionHypercholesterolemia is a risk factor for early onset of atherosclerosis in childhood.3,9
There are currently no universally accepted guidelines from the various pediatric societies on screening for hypercholesterolemia in childhood. However, there is agreement on selective screening of children with a family history of premature cardiovascular disease (before the age of 55 in men and before 65 in women) or lipid disorders, those with unknown family history or with other risk factors, particularly obesity, hypertension and diabetes,2,3,5,7,15 although selective screening will not identify all children with hypercholesterolemia.5,7 Screening should not take place before the age of two, since dietary restrictions at an earlier stage could affect the rapid growth and psychomotor development that are characteristic of the first two years of life.2,6,7 A publication in December 2011 laid out the current position of the American Academy of Pediatrics, which recommends universal screening at age 9 but highlights the need for assessment of at-risk children from the third year of life.16
The minimum age of the study population at the time of referral was two and a half years.
Of the study population, 46 presented a phenotype compatible with FH, of autosomal dominant inheritance in most cases, which results in a deficit of LDL receptors and hence reduced LDL clearance from the circulation.
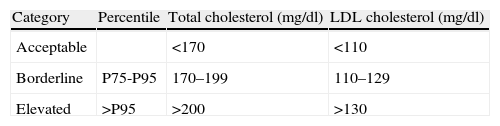
Clinical suspicions should be aroused for the diagnosis when LDL-C levels are above the 95th percentile for age and gender, and there is a family history of similar alterations in lipid profile or of premature cardiovascular disease.3 Internationally accepted cutoffs defining normal total, LDL and HDL cholesterol levels were established by the National Cholesterol Education Programme (NCEP) in 1992,17 and remained unchanged until the latest revision in 201116 (Table 4).
Cutoffs for total cholesterol and low density lipoprotein cholesterol in children and adolescents.
| Category | Percentile | Total cholesterol (mg/dl) | LDL cholesterol (mg/dl) |
| Acceptable | <170 | <110 | |
| Borderline | P75-P95 | 170–199 | 110–129 |
| Elevated | >P95 | >200 | >130 |
LDL: low density lipoprotein; P75: 75th percentile; P95: 95th percentile.
Genetic study establishes a definitive diagnosis, but is not always available.3,6,11,18 Mutations are generally identified in the LDLR gene and, less commonly, in APOB (5% of cases) or PCSK9 (2% of patients).18–20
A mutation in LDLR was identified in around 50% of our patients with FH phenotype, which is in agreement with the literature.19 No mutations were identified in APOB or PCSK9.
Assessment of lipid profile revealed significantly higher levels in the group with mutations compared to those without (total cholesterol 316.5±75.9 mg/dl vs. 260.9±42.0 mg/dl, p<0.05; non-HDL cholesterol 268.3±72.6 mg/dl vs. 203.5±43.9 mg/dl, p<0.05), as reported in other studies.13,14,20
Screening for APOE polymorphisms showed a 3.7% frequency of allele ¿2, 78.0% of allele ¿3 and 18.3% of allele ¿4. Allele ¿4 has been associated with a higher risk of coronary disease.21 As reported in other studies, individuals with allele ¿4 in our population had a stronger response to behavior modification, a finding that justified assessment of APOE polymorphisms in the present study. There appears to be no correlation between APOE polymorphisms and the efficacy of pharmacological therapy.21
The frequency of allele ¿4 was lower in individuals with a mutation in the LDLR gene (23.9% vs. 45.0%), but other studies have found no link between APOE polymorphisms and LDLR gene mutations21; the relationship found in the present study may thus be due to the small sample size.
Of the total of 168 children, 113 were prescribed behavior modification only, based on a lipid-lowering diet and exercise. The other 55 patients required pharmacological therapy as well as behavior modification in order to reduce LDL-C to acceptable levels in accordance with the latest American Academy of Pediatrics guidelines.16
Patients with FH phenotype (73.9% vs. 16.4%), and among these, those with a mutation (90.9% vs. 58.3%), more often required pharmacological intervention. This may be due to their initially higher mean levels of total and non-HDL cholesterol, as well as possibly to a weaker response to diet modification in those with FH phenotype.
Dietary changes are designed to reduce total fat intake, particularly saturated fatty acids, to 7% of total calorie intake and dietary cholesterol to 200 mg/day, with trans fatty acids making up less than 1% of total calorie intake.2,6,15,22,23 Increased fiber intake and supplementation of some dietary products with plant steroids have also been associated with improvement, albeit modest, in lipid profiles.2,3,5,22,23 Exercise is mainly reflected in increased HDL-C and reduced triglycerides, although reductions in LDL-C have also been reported.2,3,24
According to the literature, LDL-C is reduced by 10–20% through diet modification, while pharmacological therapy is generally associated with larger reductions: 10–20% for resins, 20–50% for statins, and 20% for ezetimibe.2,3,7,23. As in other studies, changes in lipid profile in our study population were more significant in those under pharmacological therapy than in those prescribed behavior modification only.
Pharm logical therapy for dyslipidemia in children is based mainly on three drug classes: resins, which act by inhibiting bile acid reabsorption in the gastrointestinal tract; statins, which decrease cholesterol production in the liver by inhibiting 3-hydroxy-3-methylglutaryl-coenzyme A reductase (responsible for endogenous cholesterol production), thus increasing LDL receptor expression and promoting LDL clearance from the circulation; and ezetimibe, which decreases intestinal absorption of dietary cholesterol.2,3,6,25,26
Fibrates, nicotinic acid and fish oil also have a role, although their use in children is limited. Fibrates are not approved for use at pediatric ages and are not used for FH in children.16 Niacin (nicotinic acid) is effective in reducing LDL-C and triglycerides by decreasing production of very low-density lipoprotein in the liver, but it is not recommended in children due to its side effects (flushing in 75% of cases, liver failure with changes in liver enzymes in 25%, myopathy, glucose intolerance and hyperuricemia).2,27 Omega-3 long-chain fatty acids found in fish oil act mainly by decreasing triglyceride synthesis in the liver and by promoting fatty-acid degradation/oxidation, increasing HDL-C but also LDL-C; they are approved for use in adults, but there have been no clinical trials in children with FH and so they are generally not used at pediatric ages.16
Recent studies have demonstrated the efficacy and safety of the available pharmacological agents, particularly in pre-pubertal children.3,23,28 Drug therapy in children aged under eight is only recommended in cases of marked elevation of LDL-C (>500 mg/dl), usually found in homozygous forms of FH.2 Children and adolescents with diabetes, kidney disease, congenital heart disease, connective tissue disease or cancer require more aggressive therapy.2
Resins, generally used in children under eight, are associated with poor compliance due to their granular and relatively insoluble presentation and the resulting gastrointestinal discomfort, particularly flatulence and constipation,2,3,5,7,15 as was found in the present study. Recently, colesevelam, a second-generation resin with greater intestinal bile acid-binding properties and improved tolerability, has attracted interest but there is little experience of its use in pediatric populations, and even then only in children aged over ten.3,5,29
The side effects usually associated with statins are myalgia and elevation of liver and muscle enzymes, occasionally associated with episodes of rhabdomyolysis, and so clinical and laboratory monitoring is mandatory.2,3,5,7,15,28 Concerns over teratogenicity mean that statins are contraindicated in pregnancy and breast-feeding.5 The safety standards observed in recent studies show that statins are indicated after the age of eight, irrespective of pubertal status, at times even as first-line therapy.2,8,15,28,30 The only side effects of ezetimibe are gastrointestinal, and it is thus useful in pediatric populations.2,15,25
The side effects of the drugs prescribed in our study were insignificant, mainly poor compliance with resin therapy due to its granular formulation, in both pre-school and school-age children; there was one case of muscle enzyme elevation, although to less than 10 times the normal level and the patient remained asymptomatic; pravastatin was discontinued as a precaution, after which enzyme levels rapidly normalized.
Pravastatin is the only statin prescribed in the pediatric nutrition unit. It is the most commonly used statin in Europe and the only one approved by the US Food and Drug Administration for children from the age of eight,2 unlike other drugs such as lovastatin, simvastatin, fluvastatin, atorvastatin and rosuvastatin that are only approved in children aged 10 and over.31 Most studies on statins at pediatric ages have been on pravastatin or lovastatin; those with pravastatin have the longest follow-up (up to four and a half years), considerably longer than those on simvastatin (maximum 12 months), rosuvastatin (maximum 40 weeks), and atorvastatin (a single study lasting 26 weeks),16 all of which showed the drugs to be safe.16,31,32 This is an important point since these patients will require lifelong therapy beginning at an age at which they are still growing and developing, and while statins have been shown to be safe in the short term, there are naturally concerns about possible adverse effects in the medium and long term. An important study with pravastatin analyzed not only its effect on lipid profile (as most other studies) but also reductions in carotid intima-media thickness, a strong marker of atherosclerotic risk.33
Since dyslipidemia is an important cardiovascular risk factor that often affects patients from childhood and into adulthood, it is essential to ensure appropriate assessment and intervention at pediatric ages, particularly of high-risk children.1
ConclusionsHypercholesterolemia is a risk factor for early onset of atherosclerosis.
A mutation in the LDLR gene was identified in around 50% of patients with FH phenotype, in agreement with the literature, and those with a mutation presented higher total and non-HDL cholesterol.
Individuals with the APOE allele ¿4 showed a stronger response to behavior modification than those without.
Improvements in lipid profile were more marked in patients prescribed pharmacological therapy as well as behavior modification, and side effects were insignificant.
It is possible to maintain the lipid profile of most individuals with FH within the normal range, and thus to reduce their risk of early development of the atherosclerotic process, which is known to be linked to serious cardiovascular complications from an early age.
At-risk children should be systematically screened from the third year of life and all others from the age of nine, in accordance with the latest guidelines, since most cases go undiagnosed, with serious consequences in terms of cardiovascular health.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data and that all the patients included in the study received sufficient information and gave their written informed consent to participate in the study.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.
AC Alves received a doctoral grant from the Portuguese Foundation for Science and Technology (SFRH/BD/27990/2006) and AM Medeiros and the Portuguese Familial Hypercholesterolemia Study received funding from the Portuguese Society of Cardiology.
Please cite this article as: Espinheira MCS, et al. Hipercolesterolemia – uma patologia com expressão desde a idade pediátrica. Rev Port Cardiol. 2013. http://dx.doi.org/10.1016/j.repc.2012.09.008.
