





Fabry disease is a rare X-linked lysosomal storage disorder caused by mutations in the alpha-galactosidase gene. The most frequent cardiac presentation of Fabry disease is cardiomyopathy characterized by left ventricular (LV) hypertrophy, usually concentric.
Heart disease in affected females tends to be clinically recognized later than in males and cardiac complications are the most frequently reported cause of death in females with Fabry disease. There are few data regarding the association between Fabry disease and LV noncompaction. We report a case of a 30-year-old asymptomatic woman, heterozygous for a nonsense alpha-galactosidase gene mutation (p.R220X), who presented LV noncompaction on cardiac magnetic resonance imaging, without LV wall hypertrophy. Histopathological examination of myocardial fragments showed marked deposition of glycosphingolipids in cardiomyocytes, confirming the diagnosis of Fabry cardiomyopathy. Based on this finding, the patient was proposed for enzyme replacement therapy. This case illustrates the role of endomyocardial biopsy in the clarification of doubtful or atypical findings related to cardiac Fabry disease, even in heterozygous women, and corroborates the contention that Fabry disease should be included in the differential diagnosis of LV hypertrabeculation/noncompaction.
A doença de Fabry é uma doença rara de armazenamento lisossómico, ligada ao cromossoma X, causada por mutações no gene da α – galactosidase. A apresentação cardíaca mais frequente é uma miocardiopatia caracterizada por hipertrofia ventricular esquerda geralmente concêntrica.
Nas mulheres afetadas a doença cardíaca tende a ser clinicamente reconhecida mais tardiamente do que em homens e as complicações cardíacas são a causa mais frequente de morte reportada em mulheres com doença de Fabry. Existem poucos dados sobre a associação entre a doença de Fabry e a não compactação do ventrículo esquerdo (VE). Reportamos o caso de uma mulher assintomática, com 30 anos de idade, heterozigota para uma mutação nonsense do gene da α–galactosidase gene (p.R220X) que apresentava critérios de VE não compactado na ressonância magnética cardíaca, sem hipertrofia das restantes paredes ventriculares. O exame histopatológico de fragmentos do miocárdio mostrou deposição acentuada de glicoesfingolípidos nos cardiomiócitos, corroborando o diagnóstico de miocardiopatia de Fabry. Com base nestes achados, foi proposto o início de terapia de substituição enzimática. Este caso ilustra o papel da biópsia endomiocárdica no esclarecimento de achados duvidosos ou atípicos relacionados com a doença de Fabry cardíaca, mesmo em mulheres heterozigotas, e corrobora a afirmação de que a doença de Fabry deve ser incluída no diagnóstico diferencial da hipertrabeculação/não compactação do VE.
Fabry disease is a rare X-linked lysosomal storage disorder caused by mutations in the alpha-galactosidase gene (GLA). Partial or complete deficiency of alpha-galactosidase enzyme activity leads to progressive intracellular accumulation of neutral glycosphingolipids containing terminal alpha-D-galactosyl residues, especially of globotriaosylceramide (Gb3), in many different cell types and tissues.1–3 The multisystemic manifestations of the classical phenotype of Fabry disease, which is typically observed in males with absent or extremely low alpha-galactosidase activity,3 include acroparesthesias and other neuropathic symptoms, angiokeratomas, hypohidrosis, gastrointestinal problems and cornea verticillata, usually beginning in childhood or adolescence, and later-onset major kidney, cardiac and cerebrovascular complications.2–4 Deacylated globotriaosylceramide, globotriaosylsphingosine (lysoGb3) may be an important pathogenic mediator involved in the onset and progression of some of the clinical and pathological manifestations of Fabry disease.5
Accumulation of Gb3 may occur in all the cellular components of the heart,3 causing a variety of clinical manifestations, including left ventricular hypertrophy (LVH), valvular disease (especially mitral regurgitation), myocardial ischemia, and arrhythmias.3 The most frequent cardiac presentation of Fabry disease is a cardiomyopathy characterized by progressive, usually concentric, LVH and replacement fibrosis with preferential localization in the basal posterolateral left ventricular (LV) wall segments.6 Heart disease in affected females tends to be clinically recognized later than in males, usually after the fourth decade of life.4,7,8 Cardiac complications are the most frequently reported cause of death in females with Fabry disease.3
Abnormalities of tissue Doppler mitral annulus velocities can be observed in patients with normal thickness of the cardiac wall, representing an early sign of myocardial damage.9
Other echocardiographic findings have been associated with Fabry disease, although none is pathognomonic. The appearance of a binary endocardial appearance,10 prominent papillary muscles or right ventricular involvement may also be encountered in ventricular hypertrophy secondary to other etiologies.11,12 There are fewer data regarding the association between LV noncompaction (LVNC) and Fabry disease.
We report a diagnosis of LVNC by cardiac magnetic resonance imaging (CMRI) in a young woman with histologically confirmed Fabry disease cardiomyopathy.
Case reportA 30-year-old woman, heterozygous for a nonsense GLA mutation (p.R220X) associated with the classical phenotype of Fabry disease, was referred to the cardiology clinic for routine screening of cardiovascular manifestations of the disease. The assay of alpha-galactosidase in leukocytes had revealed a mild deficiency of enzymatic activity (25 nmol/h/mg; normal range: 36-80), as typically observed in heterozygotes.
The patient was asymptomatic and did not manifest any other typical signs of the disease except for cornea verticillata on slit lamp ophthalmological examination, and two small angiokeratomas, on the face and in the right inframammary region. Her blood pressure, chest examination and heart and lung auscultation were normal.
Renal function was normal, with plasma creatinine level of 0.65 mg/dl and urinary albumin/creatinine ratio of 4.8 mg/g. Plasma lysoGb concentration was 7.45 nmol (normal range: 0-2.2), and the urinary excretion of Gb3 and lysoGb3 were, respectively, 36 μg/mmol of creatinine (normal range: 0-25) and 67 pmol/mmol of creatinine (normal range: undetectable).
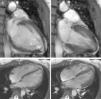
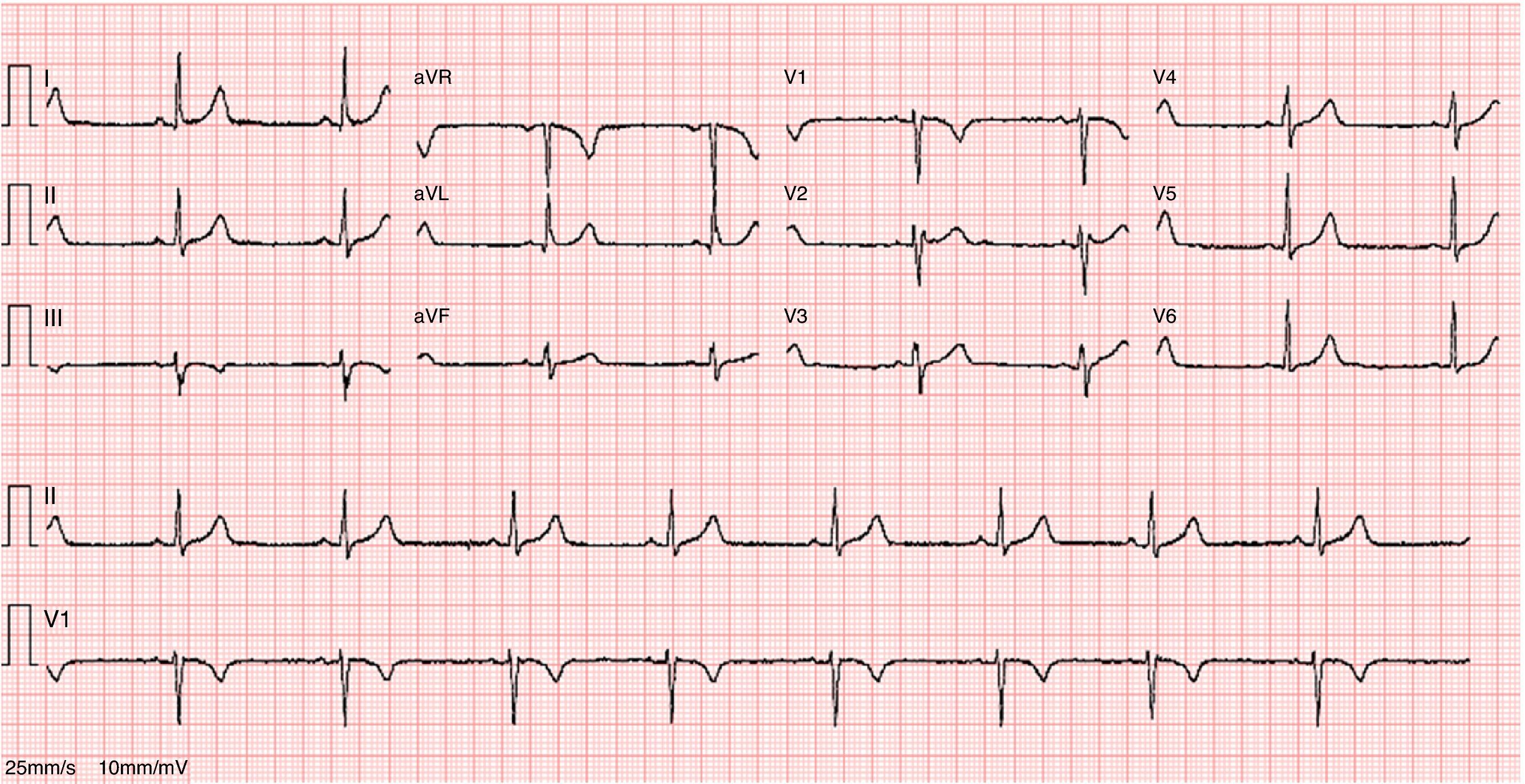
The ECG revealed sinus bradycardia, without criteria of LVH or a short PR interval (Figure 1). The 24-hour Holter study revealed no rhythm abnormalities. The echocardiogram showed normal-sized chambers and normal basal and mid LV wall thickness. Systolic and diastolic functional parameters were within normal limits, including systolic (Sa), early diastolic (Ea), and late diastolic (Aa) tissue Doppler velocities at the mitral valve annulus. The 4-chamber apical view suggested the presence of hypertrabeculation of the apical segments of the LV, but without clear criteria of noncompaction due to insufficient acoustic window quality. For this reason CMRI was performed.
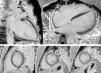
The CMRI revealed marked trabeculation in the apical segments of the LV with a diastolic ratio of noncompacted/compacted layers of 2.6, fulfilling CMRI criteria for noncompaction (Figures 2–4). LV mass was within normal limits and no delayed enhancement suggestive of fibrosis was detected after gadolinium administration.
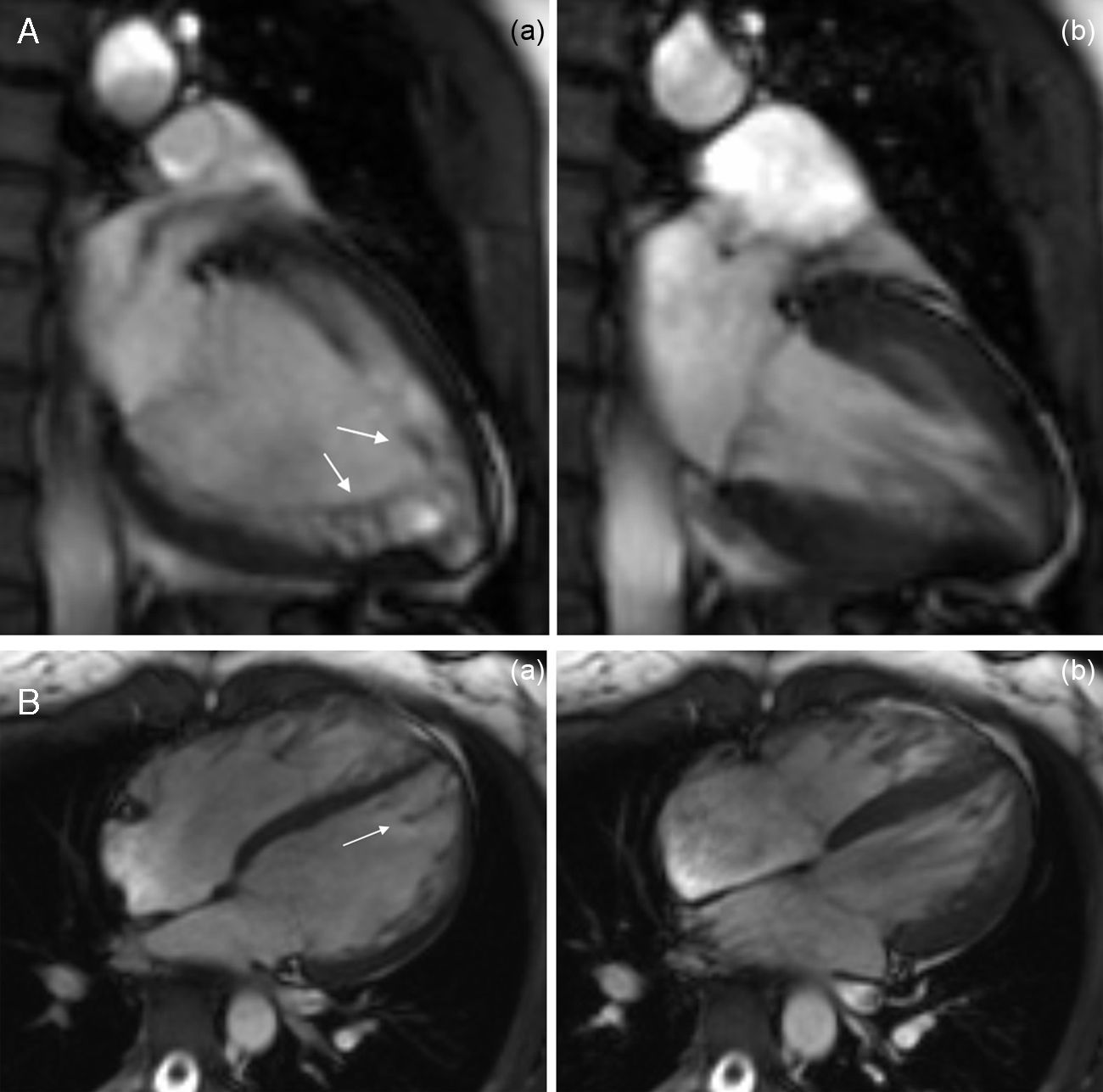
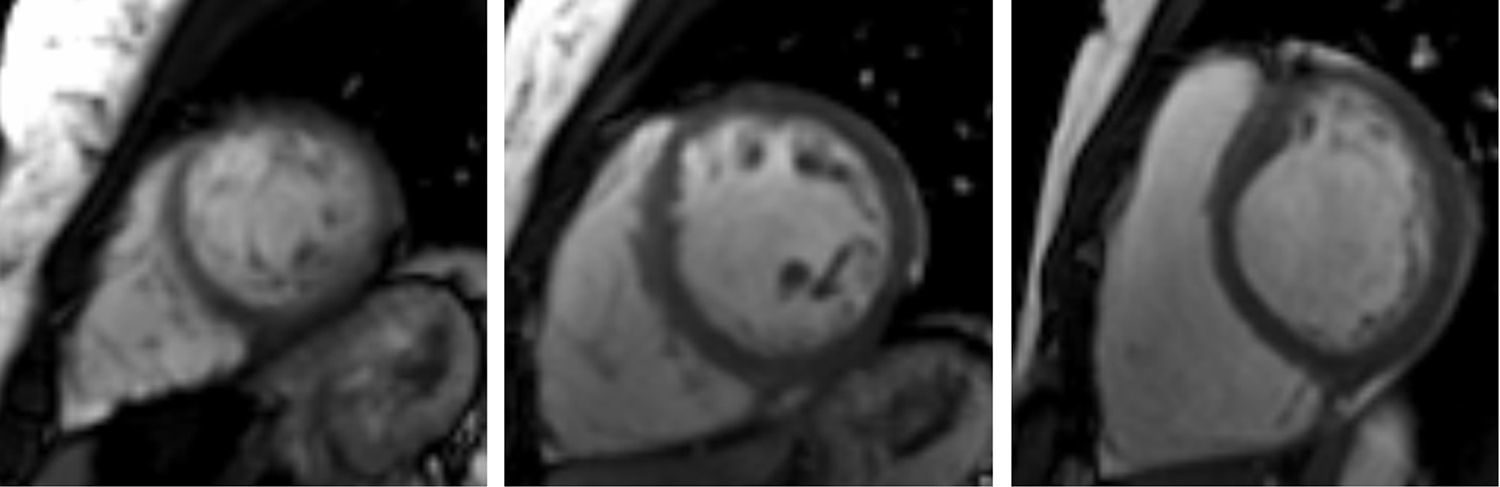
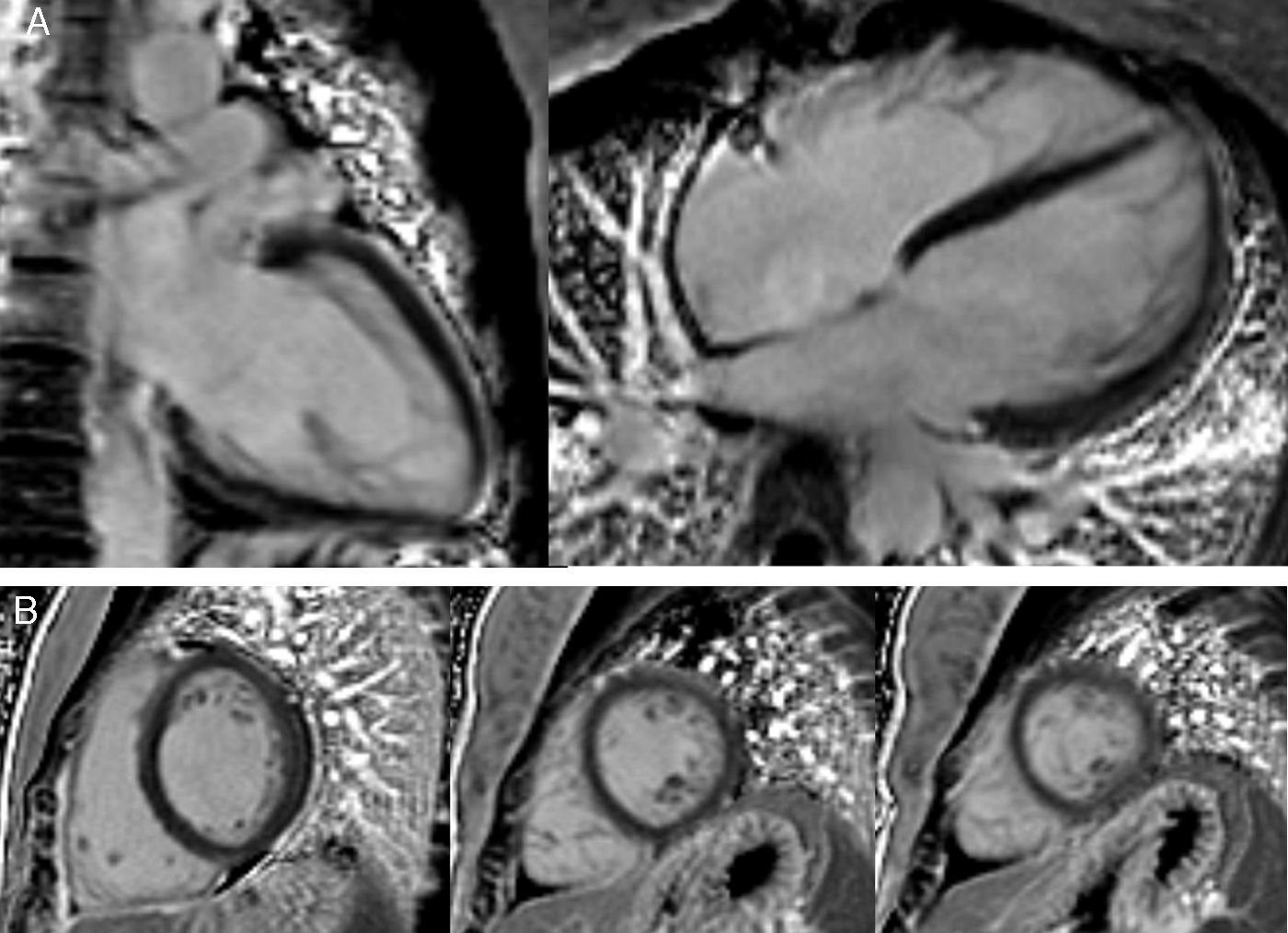
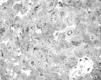
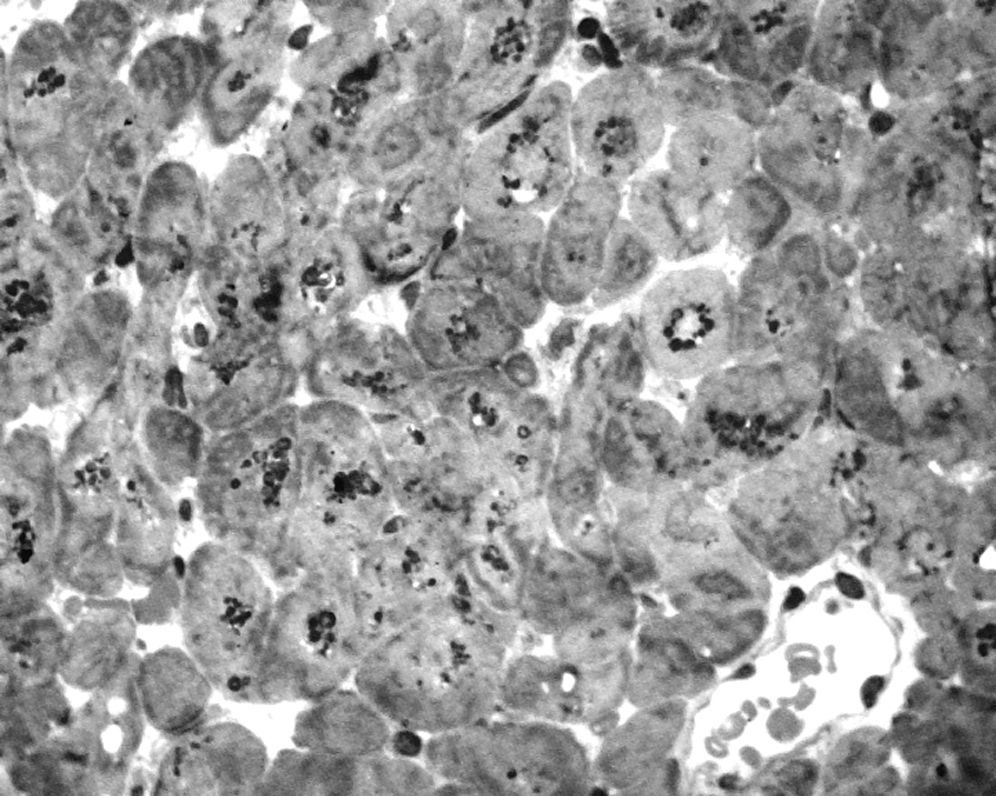
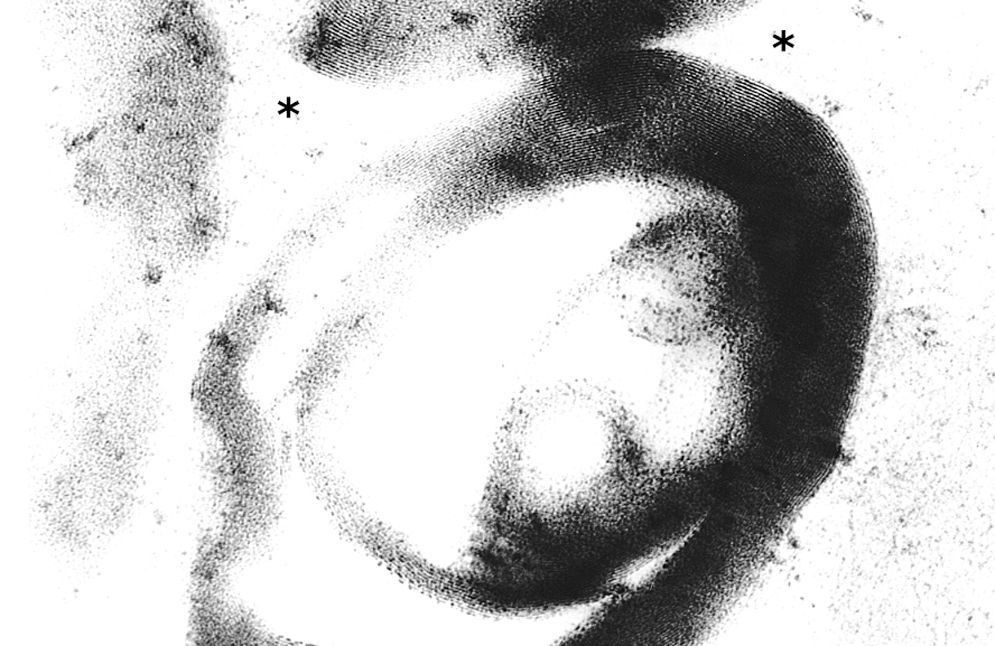
To further investigate the relationship between this morphological finding and Fabry cardiomyopathy in a female with Fabry disease and no other evidence of cardiac involvement, a transjugular endomyocardial biopsy was obtained. Histopathological examination of myocardial fragments from the right ventricle showed marked deposition of glycosphingolipids in cardiomyocytes, sparing the endothelial cells (Figures 5 and 6). Based on this finding, the patient was proposed for enzyme replacement therapy.
LVNC is a focal myocardial disorder characterized by a two-layered structure in which a noncompacted layer with numerous prominent trabeculations and deep intertrabecular recesses overlaps a thinner compacted epicardial layer.13 This morphological abnormality has been attributed to an embryonic arrest of the myocardial morphogenesis process, possibly due to abnormal regulation of cardiomyocyte polarization and myofibrillogenesis,14 but “late-onset” variants and even “reverse remodeling” of the ventricular noncompaction pattern have also been described, supporting the hypothesis that it could also just be a morphological variant of other cardiomyopathies.15–17 Given the considerable controversy surrounding its pathogenesis, diagnosis and management, the European Working Group of Myocardial Diseases considers LVNC as an unclassified form of cardiomyopathy.18
The genetic basis is heterogeneous and isolated LVNC has been described in association with various hereditary disorders, mostly due to cardiac sarcomeric protein mutations,19 but also in some myopathies and metabolic diseases.14,20–22 Reports of an association between Fabry disease and LVNC are scarce. Azevedo et al. recently described the finding of an LVNC pattern in a 32-year-old woman,23 heterozygous for the GLA mutation p.F113L, which was originally reported in association with a late-onset cardiac phenotype, with preservation of some residual enzyme activity.24 However, as these authors provided no other evidence of Fabry cardiomyopathy in their case, their suggestion that LVNC might be a rare cardiac manifestation of Fabry disease was based solely on the improbability of the patient having two rare, unrelated cardiac diseases.23 It is noteworthy that Stöllberger et al. had emphasized the importance of endomyocardial biopsy to conclusively establish an association of Fabry disease with LV hypertrabeculation/noncompaction.25
Unlike the referenced case the diagnosis of Fabry cardiomyopathy in our patient was unequivocally demonstrated by histopathological criteria. However, due to the patchy cardiac involvement expected from random X-chromosome inactivation in females heterozygous for X-linked diseases,26 it is not possible to conclude definitely that the noncompaction pattern arose in an alpha-galactosidase deficient region of the LV myocardium.
In patients with Fabry disease, irrespective of their gender, contraction and relaxation tissue Doppler mitral annulus velocities are inversely related to LV mass, but may be already significantly reduced prior to development of LVH,27 constituting an early marker of disease progression.28 The normal tissue Doppler parameters in our patient strongly suggest that Fabry cardiomyopathy was diagnosed at an early stage, still without significant disturbance of myocardial performance. Common to our patient and to the patient previously reported is the diagnosis of LVNC in young adult females with Fabry disease, at an early stage in the natural history of Fabry cardiomyopathy. As LVNC is a very unusual finding in Fabry disease,25 the remarkable similarities between the two cases are unlikely to be merely fortuitous.
The plasma of patients with Fabry disease contains a substance – possibly lysoGb35 – that stimulates proliferation of cardiomyocytes in vitro.29 In addition, studies in isolated cardiomyocytes of patients with Fabry disease have shown that Gb3 accumulation leads to degradation of myofilament proteins and to a dysfunctional state characterized by abnormally high resting tension and abnormally low active tension.30
On the other hand, a number of molecular mechanisms have been described in animal models that might contribute to the development of myocardial hypertrabeculation/noncompaction.14,31 Among these, the development of noncompaction without an increase in cell proliferative activity in trabecular LV myocardium, and the role of impaired myofibrillogenesis in the pathogenesis of noncompacted myocardium, may be particularly helpful in understanding the development of LVNC in adult patients with Fabry disease.
Taking into consideration (i) the foregoing human and animal experimental data, (ii) the rarity of LVNC in patients with Fabry disease, and (iii) the appearance, disappearance or changing morphological pattern of LVNC that have been described in several cases,32 it can be hypothesised that Fabry disease is a risk factor for myocardial noncompaction (possibly related to myofibrillolysis in cardiomyocytes) that would fully manifest only in patients carrying additional genetic or other risk factors for its development. Furthermore, expression of LVNC in Fabry disease might occur transiently and at an early stage in the natural history of Fabry cardiomyopathy. Long-term follow-up of affected patients will help to elucidate these issues.
As conclusions, our case illustrates the important role of endomyocardial biopsy in the clarification of doubtful or atypical findings related to cardiac Fabry disease, even in heterozygous women, and corroborates the contention that Fabry disease should be included in the differential diagnosis of LV hypertrabeculation/noncompaction.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Martins E, Pinho T, Carpenter S, Leite S, Garcia R, Madureira A, et al. Evidência histopatológica de doença de Fabry numa doente com não compactação do ventrículo esquerdo 2014. Rev Port Cardiol. 2014;33:565.
