




Hypertrophic cardiomyopathy is one of the main causes of sudden cardiac death in young athletes. Differentiating between this pathological condition and ‘athlete's heart’ can be quite challenging, warranting a thorough clinical and imaging assessment. Clinicians often rely on detraining-induced attenuation of electrocardiographic and echocardiographic findings as a means of distinguishing between pathological and physiological cardiac remodeling. This report describes detraining-related regression of left ventricular hypertrophy in a young soccer player with a diagnosis of hypertrophic cardiomyopathy. It challenges the dogma that regression of electrocardiographic abnormalities and left ventricular hypertrophy is exclusive to physiological remodeling and questions the impact of exercise training in the phenotypic expression and progression of hypertrophic cardiomyopathy.
A miocardiopatia hipertrófica é uma das principais causas de morte súbita de origem cardíaca em jovens atletas. Distinguir entre esta patologia e o “coração de atleta” pode ser difícil, exigindo uma avaliação clínica e imagiológica exaustiva. Frequentemente recorre-se à atenuação dos achados eletrocardiográficos e ecocardiográficos induzidos pela pausa desportiva como forma de distinguir entre remodelagem fisiológica e patológica. Neste caso clínico descrevemos a regressão da hipertrofia ventricular esquerda induzida pela pausa desportiva num jovem futebolista com o diagnóstico de miocardiopatia hipertrófica. Este caso põe em causa o dogma de que a regressão das alterações eletrocardiográficas e a da hipertrofia ventricular esquerda é exclusiva da remodelação fisiológica e, simultaneamente, levanta dúvidas sobre a influência do treino físico na evolução e expressão fenotípica da miocardiopatia hipertrófica.
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiovascular disease.1 It is characterized by a hypertrophied nondilated left ventricle in the absence of other causes.2 HCM has a highly variable phenotypic expression and is one of the most common causes of sudden cardiac death (SCD) in young athletes.3 Differentiating between HCM and the physiological remodeling seen in ‘athlete's heart’ can be quite challenging and has been the subject of controversy for years.4–8 Deconditioning has long been regarded as an important tool to distinguish athlete's heart from cardiomyopathy. The idea that regression of electrocardiographic (ECG) and/or echocardiographic abnormalities on detraining means physiological remodeling is widespread and is in fact one of the criteria proposed in the guidelines when dealing with cases that fall into the so-called gray zone of left ventricular (LV) wall thickness.9
We present a case report that may challenge this dogma: a 22-year-old male soccer player with an apical form of HCM who experienced significant regression of LV hypertrophy, as expressed by ECG and echocardiographic changes, after detraining.
Case reportA 22-year-old Caucasian male soccer player undergoing a pre-participation exam (PPE) presented a very unsettling ECG (Figure 1), highly suggestive of cardiomyopathy. He was asymptomatic, had a normal physical exam and his family history was negative for cardiovascular (CV) disease and SCD. He had been playing amateur soccer at regional level for ten years, training three days a week (90 min), with a weekly match. The transthoracic echocardiogram (TTE) revealed a nondilated left ventricle with asymmetrical apical hypertrophy (maximum LV wall thickness 16 mm) (Figure 1 and Video 1) and normal left ventricular ejection fraction and diastolic function. On cardiac magnetic resonance (CMR) the maximum apical thickness was 14 mm and the LV cavity had a spade-like appearance during diastole. No late gadolinium enhancement (LGE) was detected (Figure 2). Holter monitoring and treadmill exercise testing were normal (16 metabolic equivalents, no symptoms and no arrhythmias) and genetic testing was negative for the following mutations: MYH7, MYBPC3, ACTC1, TNNT2, TNNI3, TPM1, MYL2, MYL3, CSRP3 and TCAP.
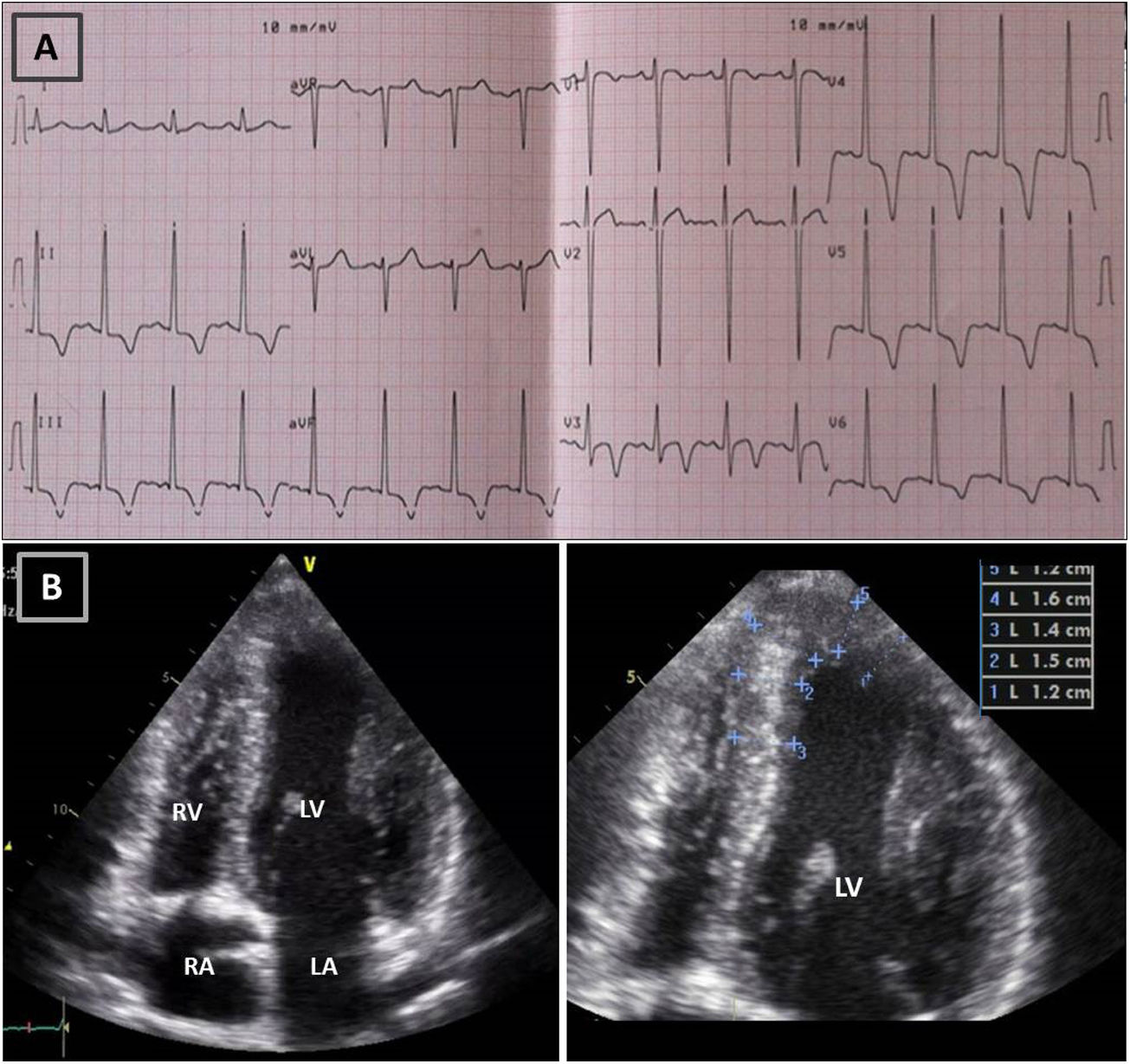
Resting electrocardiogram (ECG) (top) and transthoracic echocardiogram (TTE) (bottom), before detraining. (A) ECG with voltage criteria for LV hypertrophy and marked deep T-wave inversion in leads I, II, III, aVF and V3-V6, with ST-segment depression; (B) TTE (apical 4-chamber view), showing moderate asymmetric hypertrophy, localized at the mid-apical segments of the left ventricle, with maximum wall thickness of 16 mm. LA: left atrium; LV: left ventricle; RA: right atrium; RV: right ventricle.
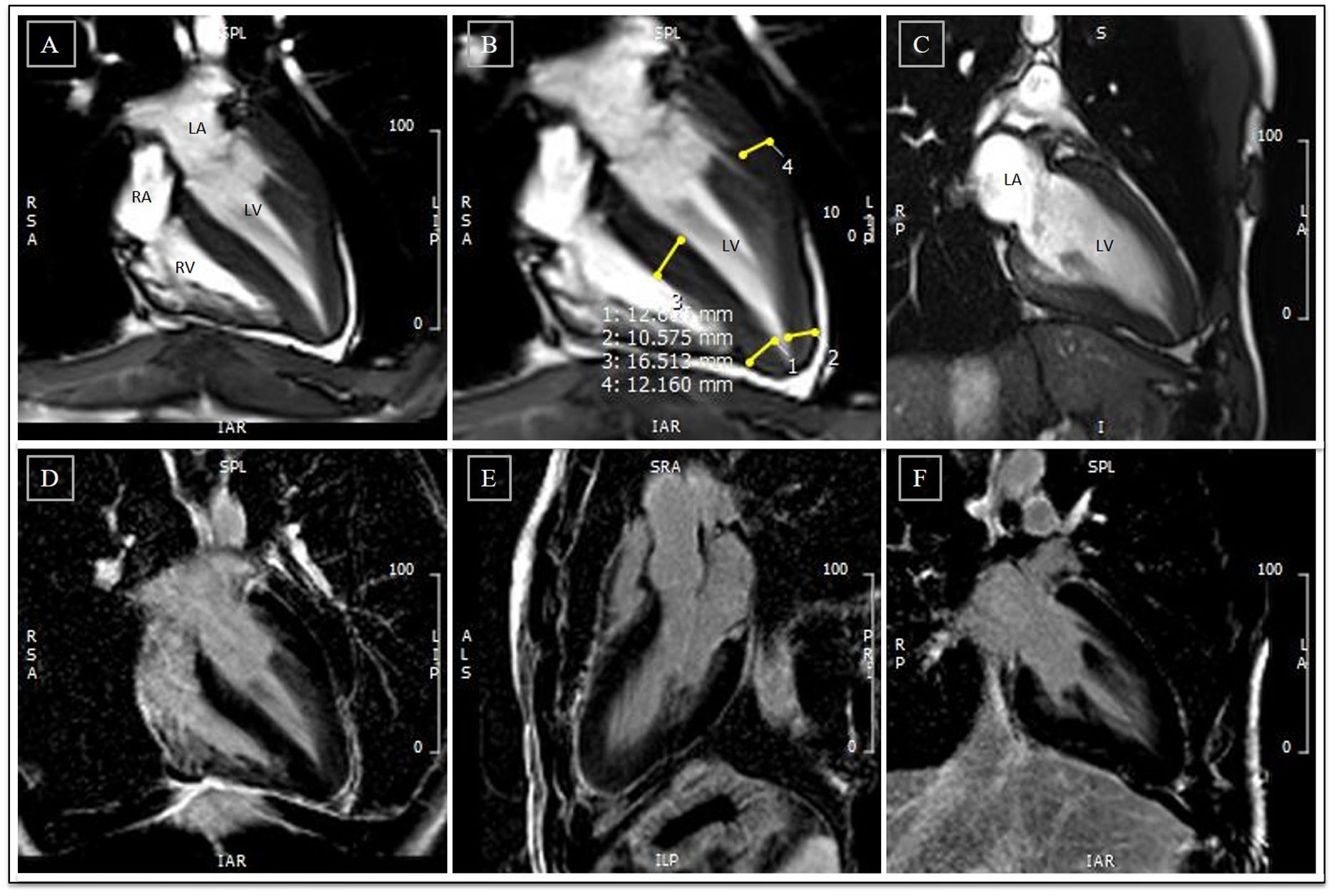
Cardiac magnetic resonance before detraining. (A-C) Cine steady state free precession sequences (in diastole) with overlapping echocardiographic findings – similar wall thicknesses, particularly in the LV apical segments. Long-axis horizontal views enable visualization of the apex, with a clearly abnormal cavity morphology, confirming the diagnosis of apical hypertrophic cardiomyopathy, mixed form, with an ‘ace of spades’ configuration; (D-F) delayed enhancement sequences showing absence of myocardial fibrosis. LA: left atrium; LV: left ventricle; RA: right atrium; RV: right ventricle.
Taken together, these findings were compatible with an apical form of HCM and he was advised against participation in competitive sports.
Follow-up assessment was performed four months later. He had reportedly respected our warning and barely exercised in the meantime. While still abnormal, the ST-segment depression and deeply inverted T waves had become less prominent (Figure 3). We repeated the TTE and had a similar impression: although maximum wall thickness in the apex was significantly decreased (10-11 mm) and not diagnostic of HCM, the TTE seemed clearly abnormal, given the small cavity, the abnormal geometry and the apical gradient (Figures 3 and 4 and Video 2).
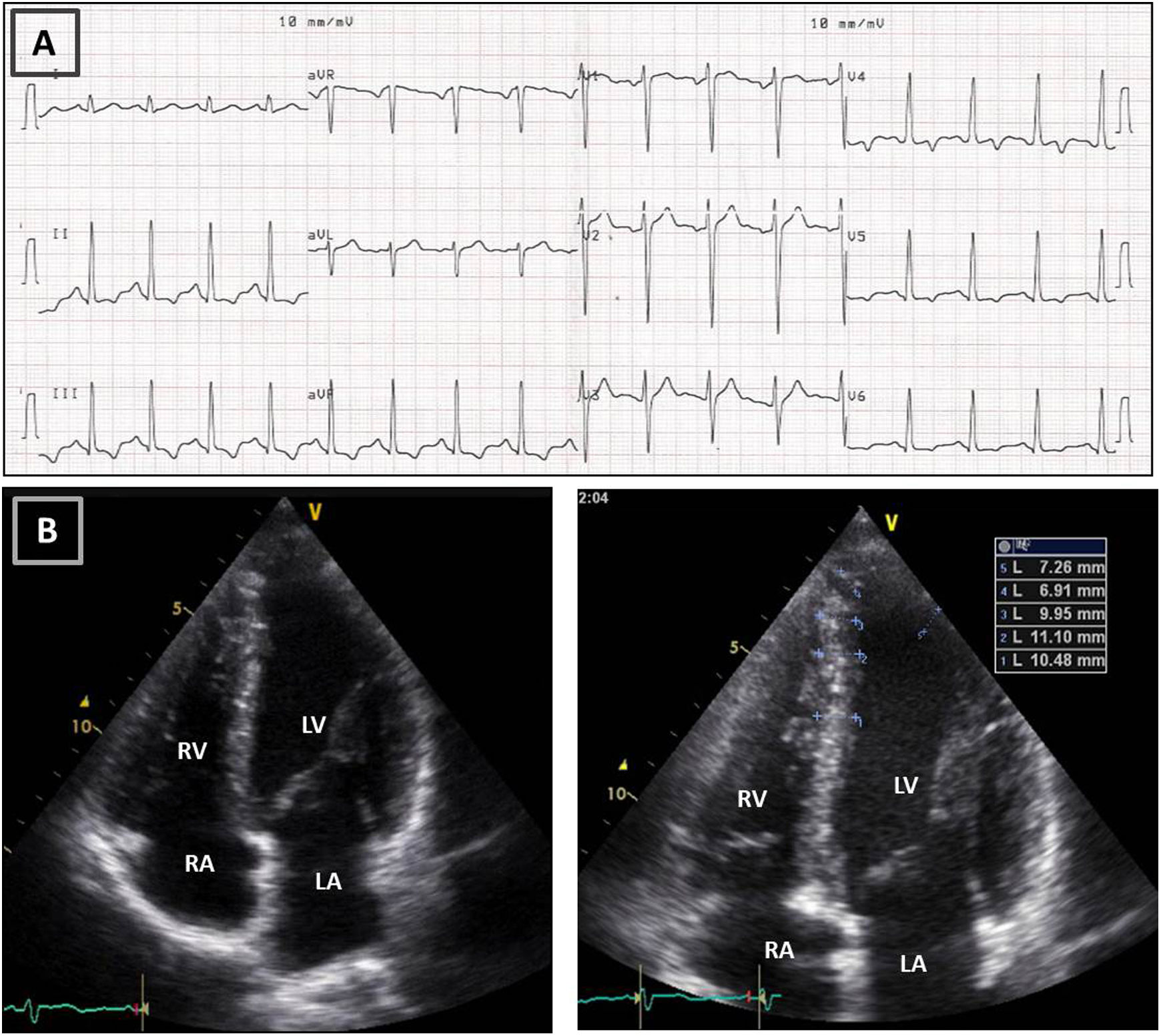
Resting electrocardiogram (ECG) (top) and transthoracic echocardiogram (TTE) (bottom), performed four months after detraining. (A) The ECG shows a clear improvement of the T-wave abnormalities in the inferior and lateral leads, although the ECG is still abnormal; (B) on TTE, a decrease in both septal and apical LV wall thickness is evident (maximum 11 mm). LA: left atrium; LV: left ventricle; RA: right atrium; RV: right ventricle.
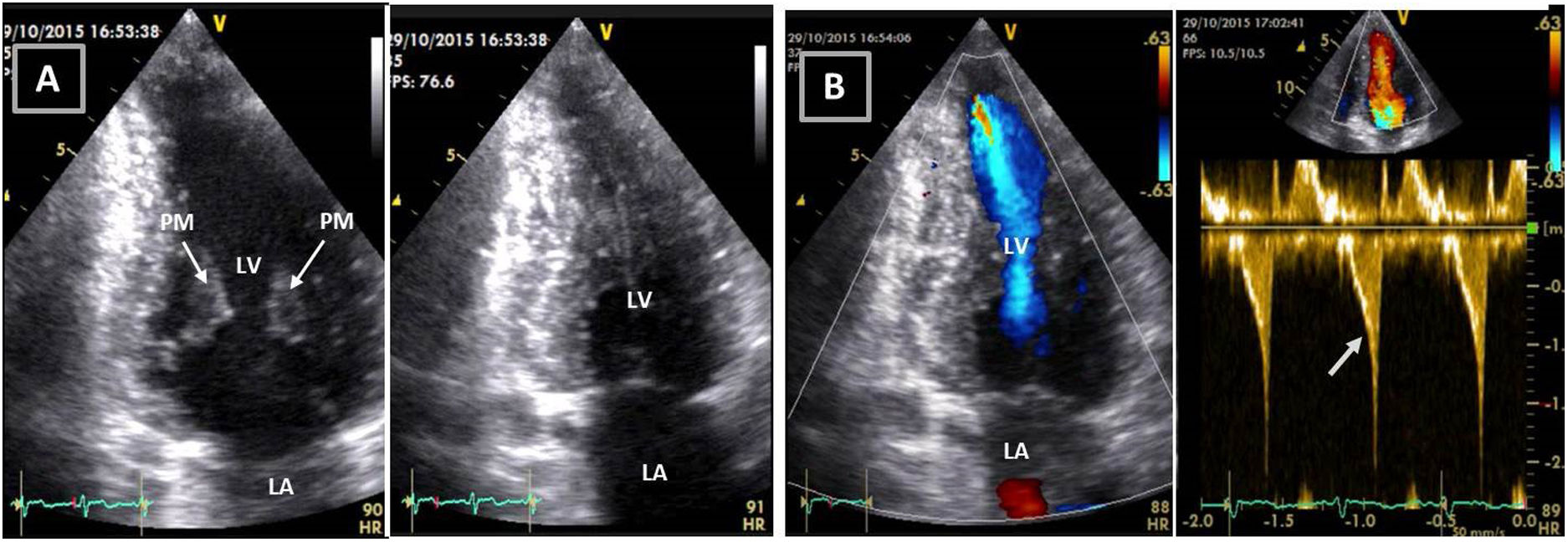
Transthoracic echocardiogram after detraining. Note the abnormal shape of the left ventricular cavity (small cavity size, prominent papillary muscle, with apical cavity obliteration in systole (A) and increased flow velocity (arrow) (B) suggestive of a myopathic left ventricle, rather than adaptive remodeling following training. LA: left atrium; LV: left ventricle; RA: right atrium; RV: right ventricle.
These ECG abnormalities had already been detected two years before our ‘PPE’ and led to a proposal for a deconditioning period. We had access to these ECGs and the pattern of pseudo-normalization was the same as shown in Figures 1 and 3.
The recommendation not to participate in competitive sports was reinforced and the young man is asymptomatic and free of CV events to date.
DiscussionIn this case giant T-wave inversions (TWI) prompted secondary investigation. The prevalence of ECG repolarization abnormalities among athletes has been reported to be 3-5%.10 TWI in the inferior and lateral leads have been identified in 1.5-1.8% of adult Caucasian athletes, while TWI in the lateral leads have been identified in only 0.3%.10 Some studies have shed light on the long-term outcome of apparently healthy athletes with marked repolarization abnormalities. Pelliccia et al. identified 81 subjects (from a database of 12 550 trained athletes) with marked repolarization abnormalities and no apparent cardiac disease who underwent serial clinical, ECG and TTE assessment for 9±7 years. Of the 81 athletes, a diagnosis of cardiomyopathy was made in five (6%).11 Papadakis et al. followed 1243 athletes for 69.7±29.6 months, together with 52 black patients with confirmed HCM. Most of the patients with HCM (77%) and the three athletes eventually diagnosed with HCM presented deep TWI in the lateral leads.12 Recently a study conducted in 2261 peri-pubertal athletes found 136 (6%) to have TWI. Of the group of five presenting TWI in the inferolateral leads, three (60%) had significant TTE abnormalities, including one case of HCM and two of LV hypertrophy.13 Schnell et al. recently published the results of 155 athletes with TWI in whom an identifiable cardiac disease was demonstrated in 44.5%.14 These and other studies support the conclusion of an editorial by Wilson and Carré in which they state that the vast majority of the evidence implicates TWI as part of a disease process, with a normal cardiac phenotype that may later develop into pathology. Accordingly, TWI in the inferolateral leads are not usually training-related and should be considered pathological until proved otherwise. Those athletes with TWI that are asymptomatic and have normal cardiac evaluation should undergo regular cardiac assessment to monitor possible disease expression.6
The current diagnostic criteria for HCM in adult patients are based on the presence of LV end-diastolic thickness of ≥15 mm in one or more myocardial segments.9,15 In a recent review,1 Sen-Chowdhry et al. pointed out that these LV hypertrophy-based criteria are not adjusted for age, gender or body surface area, and do not consider normal regional variations in LV dimensions. Moreover, they fail to identify pathology in a large number of patients whose hearts are macroscopically normal (with wall thickness and cavity size within normal limits) but already present widespread myocardial disarray on histological analysis.1 Evidence is emerging that some forms of HCM may not be captured by the existing diagnostic criteria, partially driven by the widespread use of CMR. Flett et al. reported on 22 patients who had TWI (alongside other features consistent with HCM) and relative apical thickening (apical:basal wall thickness ratio>1) but not LV wall thickness >15 mm. Given that normal LV wall thickness decreases from base to apex, they proposed that this cohort represented an extension of the apical HCM phenotype and that relative but not absolute apical hypertrophy should be used for the diagnosis of apical HCM.16 These conclusions are supported by Wu et al. in a recent study, in which they prospectively enrolled 60 otherwise healthy subjects with giant TWI and found that the apical thickness (measured on CMR) of subjects with giant TWI was significantly greater than that of controls (8.10±1.67 mm vs. 4.14±1.17 mm in males). Additionally, the apical morphology of subjects with giant TWI was significantly different from normal subjects, raising the question whether this cohort should be included within the scope of preclinical apical HCM.17 It should also be borne in mind that disease penetrance in HCM is age-dependent18 and in this particular case we may not yet be seeing full-blown LV hypertrophy.
In conferences all over the world, authors have been questioning the widely held assumption that normalization (or regression) of ECG and/or echocardiographic abnormalities after detraining means that such features may be regarded as physiological. Given that there are no prospective or retrospective trials testing this hypothesis, their belief is based on evidence arising from animal studies and from a few isolated case reports. In an animal model of HCM, Kazmierczak et al. showed that strenuous exercise can trigger hypertrophy and pathological cardiac remodeling. While such changes were not seen in >6-month-old sedentary mice, exercised mice displayed them as early as three months old.19 Kebed et al.3 published a case report of an 18-year-old hockey player with an abnormal ECG and apical HCM with maximum wall thickness (MWT) of 24 mm on CMR. He discontinued athletic training and 14 months after the initial diagnosis CMR showed MWT of 10 mm at the apex. The areas of LGE seen on the initial CMR were unchanged and, although still abnormal, the ECG changes were less prominent. He was genotype positive. The authors proposed that this patient had both athletic adaptation to training and HCM. The former had regressed while the latter was responsible for the abnormalities still observed after detraining.3 A similar case was reported by De Gregorio et al.20 A 16-year-old male soccer player with deep TWI in the inferolateral wall was diagnosed with HCM after TTE showed LV hypertrophy involving both the septum (12.4 mm) and the apex (15.8 mm). Mild diastolic dysfunction was revealed on TTE and CMR confirmed LV hypertrophy (anterior septum 13.5 mm, apical wall 14 mm, other segments 12.5-13 mm), with no evidence of LGE. He was disqualified from competitive sports and follow-up was scheduled. Six months later, the ECG findings had improved notably with deconditioning and a decrease in both septal (12.1 mm) and apical (13 mm) LV wall thickness was evident in the TTE. Once again, the athlete was discouraged from intense physical training. However, two months later, before coming for a 12-month check-up, he started playing recreational tennis. As a result, LV wall thickness had increased again (apical thickness of 14.9 mm), and longitudinal deformation, wall synchrony and ECG appearance had also worsened.20
These two cases closely resemble the patient we present here. We hypothesize that they belong to a group of patients with HCM in whom disease penetrance (and phenotype expression) may be triggered or accelerated by exercise training. Conversely, detraining has the opposite effect. In this group of patients, at any given moment, imaging and ECG findings reflect the sum of both physiological and pathological adaptations.
That being said, how should we interpret the detraining response? It can be summarized in two sentences: absence of response to detraining suggests pathological LV hypertrophy; and reduction of LV wall thickness may be observed in physiological hypertrophy but also in athletes with HCM, and does not necessarily mean athlete's heart. It is worth mentioning that athletes advised not to compete often do not comply and keep training, making the interpretation of follow-up exams very difficult.
Decision-making concerning eligibility for competitive sports can be very difficult and is the subject of controversy. The risk of adverse cardiac events and SCD is believed to be low in apical HCM. However, it is not known to what extent exercise can accelerate phenotype progression or even act as a trigger of malignant arrhythmias. Accordingly, the current guidelines advise against competitive sport participation in HCM with phenotypic abnormalities.21
In our opinion the case reported here corresponds to apical HCM with subclinical expression whose phenotype is aggravated by exercise training. Despite the absence of fully expressed disease, after discussion of the risks, the athlete was discouraged from participating in competitive sports.
ConclusionEvidence that implicates deep inferolateral TWI as part of a disease process continues to emerge. These deep TWI may represent incomplete phenotype expression, often preceding pathological abnormalities in imaging modalities. As such, athletes with TWI and a normal cardiac assessment should undergo regular cardiac monitoring with comprehensive clinical and imaging assessment, to screen for possible disease expression and provide a definitive diagnosis of HCM. Moreover, we believe that continued clinical and ECG/TTE surveillance is warranted in athletes with significant LV hypertrophy, even when initial assessment and response to detraining suggest athlete's heart.
An important conclusion that can be drawn from our case report (and the others presented above) is that we cannot blindly trust in detraining-induced attenuation of ECG and TTE changes as a marker of benign physiological adaptation. Complete or partial regression of these changes may be observed in physiological hypertrophy but also in athletes with cardiomyopathy, reflecting the complex interaction between genetic and environmental factors.
The current guidelines are still very restrictive concerning eligibility for competitive sports. New evidence and use of shared decision models may change the approach to such low-risk cases in the near future.
Conflicts of interestThe authors have no conflicts of interest to declare.
