



We report the results of the Portuguese Registry of Hypertrophic Cardiomyopathy, an initiative that reflects the current spectrum of cardiology centers throughout the territory of Portugal.
MethodsA direct invitation to participate was sent to cardiology departments. Baseline and outcome data were collected.
ResultsA total of 29 centers participated and 1042 patients were recruited. Four centers recruited 49% of the patients, of whom 59% were male, and mean age at diagnosis was 53±16 years. Hypertrophic cardiomyopathy (HCM) was identified as familial in 33%. The major reason for diagnosis was symptoms (53%). HCM was obstructive in 35% of cases and genetic testing was performed in 51%. Invasive septal reduction therapy was offered to 8% (23% of obstructive patients). Most patients (84%) had an estimated five-year risk of sudden death of <6%. Thirteen percent received an implantable cardioverter-defibrillator. After a median follow-up of 3.3 years (interquartile range [P25-P75] 1.3-6.5 years), 31% were asymptomatic. All-cause mortality was 1.19%/year and cardiovascular mortality 0.65%/year. The incidence of heart failure-related death was 0.25%/year, of sudden cardiac death 0.22%/year and of stroke-related death 0.04%/year. Heart failure-related death plus heart transplantation occurred in 0.27%/year and sudden cardiac death plus equivalents occurred in 0.53%/year.
ConclusionsContemporary HCM in Portugal is characterized by relatively advanced age at diagnosis, and a high proportion of invasive treatment of obstructive forms. Long-term mortality is low; heart failure is the most common cause of death followed by sudden cardiac death. However, the burden of morbidity remains considerable, emphasizing the need for disease-specific treatments that impact the natural history of the disease.
Apresentação dos resultados do Registo Português de Miocardiopatia Hipertrófica.
MetodologiaConvite direto aos diferentes centros de cardiologia de Portugal, com análise de dados basais e de seguimento.
ResultadosForam 29 os centros participantes e 1042 doentes incluídos. Quatro centros incluíram 49% dos doentes, 59% do sexo masculino, idade média de diagnóstico 53 ± 16 anos. A doença foi considerada familiar em 33% e a presença de sintomas foi a principal causa de diagnóstico (53%). A miocardiopatia hipertrófica foi obstrutiva em 35%. O estudo genético foi efetuado em 51%. Oito por cento dos doentes fizeram terapêutica invasiva de redução septal (23% dos doentes com obstrução). A maioria dos doentes (84%) apresentava um risco estimado de morte súbita aos 5 anos < 6%. Em 13% foi colocado desfibrilhador cardioversor implantável. Após um seguimento de 3,3 anos, intervalo interquartil (P25-P75) 1,3–6,5 anos, 31% estavam assintomáticos. A mortalidade total foi de 1,19%/ano e a cardiovascular de 0,65%/ano. A incidência de morte por insuficiência cardiaca foi de 0,25%/ano, a de morte súbita de 0,22%/ano e a de morte por acidente vascular cerebal de 0,04%/ano. A mortalidade por insuficiência cardíaca e transplante cardíaco foi de 0,27%/ano e a de morte súbita e equivalentes de 0,53%/ano.
ConclusõesA miocardiopatia hipertrófica em Portugal apresenta idade de diagnóstico elevada e é frequente o tratamento invasivo de formas obstrutivas. A mortalidade é baixa, a insuficiência cardíaca é a principal causa de morte, seguida pela morte súbita. A doença apresenta elevada morbilidade, realça a necessidade do desenvolvimento de tratamentos específicos com impacto na sua história natural.
atrial fibrillation
alcohol septal ablation
cardiac magnetic resonance
case report form
hypertrophic cardiomyopathy
heart failure
implantable cardioverter-defibrillator
interventricular septum
left ventricular hypertrophy
Portuguese Registry of Hypertrophic Cardiomyopathy
sudden cardiac death
transient ischemic attack
Hypertrophic cardiomyopathy (HCM) represents an important health burden as a cause of sudden cardiac death (SCD), heart failure (HF), atrial fibrillation (AF) and stroke. HCM shares many disadvantages of rare diseases, including limited recognition, lack of prospective studies assessing treatment, and little or delayed access to advanced treatment options without enjoying their regulatory benefits.1–4 Randomized clinical trials are infrequent in HCM and recommendations are largely based on expert consensus.1–4 Additionally, the majority of studies still originate from tertiary referral centers, and little is known about the clinical profile and management of the disease at a nationwide level. The real impact of genetics and imaging techniques on earlier and wider recognition of HCM, as well as of advanced treatment options on outcomes, is also unknown. It is of paramount importance to capture these changes and to provide answers to these questions.1,2,5
Accordingly, the importance of clinical registries of HCM is increasing, since they provide the best source of real-world data in specific countries and geographical regions.
Assuming a prevalence of 1:5005 for HCM in general and of 1:3200 for ‘clinical HCM’6 (patients who come to medical attention), the number of patients with HCM in Portugal (population about 10 million) is respectively around 200007 and 3000.6 However, few studies have addressed this population.8,9 Besides its relevance to national cardiologists, the Portuguese HCM population represents an interesting sample because of the country's relatively small size, homogeneous population and high penetration of health care.
The Portuguese Registry of Hypertrophic Cardiomyopathy (PRo-HCM) was instituted to collect information on the actual situation of HCM in Portugal. It specifically assessed epidemiological, sociodemographic and clinical data, current standards for diagnosis, treatment, follow-up, and outcomes. Another aim was to develop a reliable source of information for health professionals, patients and families, on appropriateness, effectiveness and quality of care.
MethodsRegistry design and methodologyThe PRo-HCM registry was conceived by the Working Group on Myocardial and Pericardial Diseases of the Portuguese Society of Cardiology, directed by an executive and a scientific committee, and managed in the Portuguese National Center for Data Collection in Cardiology (CNCDC). This study was formulated and conducted in compliance with the principles of the declaration of Helsinki, and approved by the National Center for Data Protection. It was an observational, multicenter, voluntary, non-mandatory study, with a two-year enrollment period (April 2013-April 2015), retrospective but including a prospective update.
A direct invitation was made to cardiology departments nationwide, central and regional, public and private, academic and non-academic, covering rural and urban, coastal and inland areas. Additionally, the registry was advertised in the Portuguese Journal of Cardiology, meetings and newsletters. If the invitation was accepted, the principal investigator received detailed instructions, a center identification number and a unique username and password to gain access to the electronic case report form (CRF) (http://www.spc.pt/RegistosMiocardiopatia/Public/Login.aspx?ReturnUrl=%2fRegistosMiocardiopatia%2f). The CRF contained seven sections: (1) patient identification and demographic/epidemiological data; (2) past history and baseline clinical data; (3) mortality and risk stratification; (4) diagnostic tests; (5) genetic testing, family screening, and genetic counseling; (6) treatment; (7) last assessment (clinical course, follow-up, and outcomes). In the diagnostic tests section the investigators were asked to enter the exams performed at the time of first assessment, including electrocardiogram (ECG), echocardiogram, ambulatory ECG, exercise test, exercise echocardiogram, cardiac magnetic resonance (CMR), and cardiac computed tomography.
Centers were asked to include all patients with a diagnosis of HCM followed at the center currently or in the past (no retrospective time limit), including those already deceased at the time of enrollment. Written informed consent was obtained from living patients and from a proxy of deceased patients.
Inclusion criteria were age >18 years at the time of enrollment, and unexplained left ventricular hypertrophy (LVH): wall thickness ≥15 mm by imaging techniques (in first-degree relatives10 ≥14 mm in the inferior interventricular septum (IVS) or lateral wall or ≥13 mm in the anterior IVS or inferior wall).
Exclusion criteria were secondary LVH (grade ≥2 hypertension11), moderate or severe aortic stenosis,12 previously diagnosed cardiac or systemic disease, and metabolic or multi-organ syndrome associated with LVH.
After the inclusion period, extra time was provided to complete the CRFs and to clean the database. The final date of registry closure was December 31, 2015. CRFs were reviewed to confirm consistency of data. Whenever necessary, queries were sent to investigators. In the event of repeated patients (same initials, gender and birth date), the one with the longer follow-up time was included.
DefinitionsThroughout the study, most data are relative to the time of first visit. When clinically relevant, data at the time of diagnosis of HCM are also shown.
Follow-up time was defined as time from initial assessment at the center to last assessment or death.
Sudden cardiac death (SCD) was defined as unexpected death occurring within one hour of symptom onset in patients who had previously experienced a relatively stable or uneventful clinical course. Resuscitation from cardiac arrest or appropriate implantable cardioverter-defibrillator (ICD) therapies for primary prevention were considered as equivalents of SCD.
HF-related death was defined as that occurring in the context of progressive cardiac decompensation, with decline in left ventricular function.13 Heart transplantation was considered as equivalent to HF-related death.
Stroke-related deaths in the setting of paroxysmal, persistent or permanent AF were classified as AF-stroke related deaths. Stroke-related deaths in the absence of documented AF were not included in this group.
Thromboembolic events, defined as stroke, transient ischemic attack (TIA) or systemic peripheral embolism, were recorded.14
The classification of identified genetic variants was assigned to the investigators, as pathogenic/probably pathogenic, of unknown significance or benign/probably benign, according to current knowledge of their pathogenicity,15,16 as provided by genetic laboratories (these data were not centrally reviewed or corrected by the coordinators of the registry). A genetic study was defined as negative if no pathogenic/probably pathogenic mutation was detected and as in progress if no result was provided at inclusion.
Statistical analysisContinuous variables were expressed as mean and standard deviation or as median and interquartile range (IQR) (P25-P75). Categorical variables were given as total number and percentages. Chi-square or Fisher tests were used for comparisons of categorical variables and Student's t tests for continuous variables. Survival was assessed by Cox proportional hazard regression. Survival curves were constructed according to the Kaplan-Meier method, and comparisons were performed using the log-rank test. All p-values were two-sided and considered significant when <0.05. All analyses were performed using SPSS 19.0®.
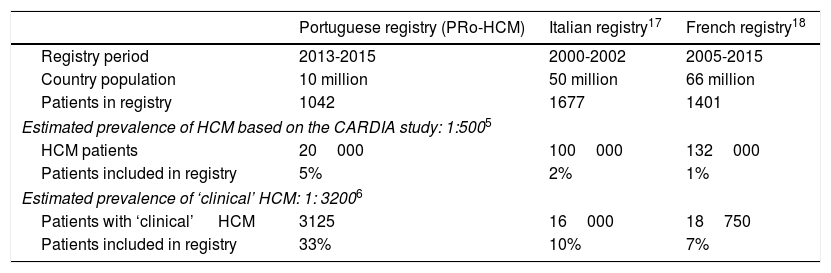
ResultsOf the 62 institutions contacted, 37 accepted, and the final number of participating centers was 29 (Figure 1). The total number of patients was 1042. Figures were compared with other national registries17,18 (Table 1).
 HCM registry: distribution by regions and by center. Left: participating centers (n=29); top right: distribution of the 1042 patients by regions of Portugal: the Lisbon region included the highest number of patients and the central region of Portugal the lowest; bottom right: note the heterogeneity in terms of patients enrolled per center.' title='Participating centers in the Pro-
HCM registry: distribution by regions and by center. Left: participating centers (n=29); top right: distribution of the 1042 patients by regions of Portugal: the Lisbon region included the highest number of patients and the central region of Portugal the lowest; bottom right: note the heterogeneity in terms of patients enrolled per center.' title='Participating centers in the Pro-Participating centers in the Pro-HCM registry: distribution by regions and by center. Left: participating centers (n=29); top right: distribution of the 1042 patients by regions of Portugal: the Lisbon region included the highest number of patients and the central region of Portugal the lowest; bottom right: note the heterogeneity in terms of patients enrolled per center.
Comparison between populations of national registries of hypertrophic cardiomyopathy.
| Portuguese registry (PRo-HCM) | Italian registry17 | French registry18 | |
|---|---|---|---|
| Registry period | 2013-2015 | 2000-2002 | 2005-2015 |
| Country population | 10 million | 50 million | 66 million |
| Patients in registry | 1042 | 1677 | 1401 |
| Estimated prevalence of HCM based on the CARDIA study: 1:5005 | |||
| HCM patients | 20000 | 100000 | 132000 |
| Patients included in registry | 5% | 2% | 1% |
| Estimated prevalence of ‘clinical’ HCM: 1: 32006 | |||
| Patients with ‘clinical’ HCM | 3125 | 16000 | 18750 |
| Patients included in registry | 33% | 10% | 7% |
HCM: hypertrophic cardiomyopathy.
Almost half of the patients (n=514, 49%) came from the four major centers with specific interest in HCM (the other 25 centers enrolled 528 patients, 51%) (Figure 1). The Lisbon region included the largest number, followed by the North region, the South and Islands, and the Central region. Of the 29 centers, only three included more than 100 patients and eight more than 50 patients. Twenty-one centers included fewer than 50 patients each, 13 centers fewer than 10 and six centers fewer than five.
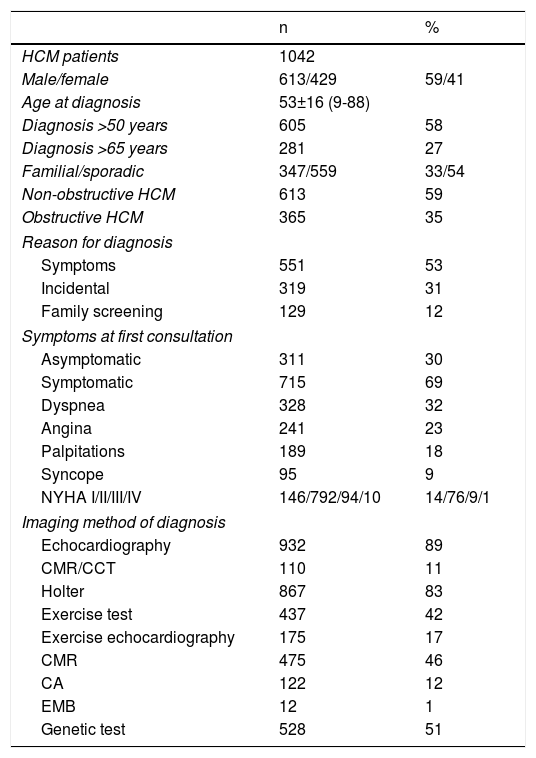
The patient cohort showed a slight preponderance of males. Mean age at diagnosis was 53±16 years and more than one quarter were diagnosed over the age of 65 years. The disease was classified as familial in one third. At first consultation most patients were symptomatic19 (Table 2).
Summary of baseline characteristics and diagnostic tests.
| n | % | |
|---|---|---|
| HCM patients | 1042 | |
| Male/female | 613/429 | 59/41 |
| Age at diagnosis | 53±16 (9-88) | |
| Diagnosis >50 years | 605 | 58 |
| Diagnosis >65 years | 281 | 27 |
| Familial/sporadic | 347/559 | 33/54 |
| Non-obstructive HCM | 613 | 59 |
| Obstructive HCM | 365 | 35 |
| Reason for diagnosis | ||
| Symptoms | 551 | 53 |
| Incidental | 319 | 31 |
| Family screening | 129 | 12 |
| Symptoms at first consultation | ||
| Asymptomatic | 311 | 30 |
| Symptomatic | 715 | 69 |
| Dyspnea | 328 | 32 |
| Angina | 241 | 23 |
| Palpitations | 189 | 18 |
| Syncope | 95 | 9 |
| NYHA I/II/III/IV | 146/792/94/10 | 14/76/9/1 |
| Imaging method of diagnosis | ||
| Echocardiography | 932 | 89 |
| CMR/CCT | 110 | 11 |
| Holter | 867 | 83 |
| Exercise test | 437 | 42 |
| Exercise echocardiography | 175 | 17 |
| CMR | 475 | 46 |
| CA | 122 | 12 |
| EMB | 12 | 1 |
| Genetic test | 528 | 51 |
CA: cardiac angiography; CCT: cardiac computed tomography; CMR: cardiac magnetic resonance; EMB: endomyocardial biopsy; HCM: hypertrophic cardiomyopathy; NYHA: New York Heart Association functional class.
The ECG was abnormal in 964 individuals (93%). AF was recorded in 117 (11%).
Echocardiographic assessment at enrollment showed that HCM was non-obstructive (instantaneous peak Doppler intraventricular pressure gradient <30 mmHg) in 613 (59% of patients) and obstructive in 365 (35%) (Table 2). Of these, 323 (88%) had obstruction at rest and 42 (12%) had exercise-induced obstruction only, during exercise echocardiography. Obstruction was at the left ventricular outflow tract in 89%. An apical aneurysm was present in 23 patients (2%).
On ambulatory Holter ECG monitoring, AF was present in 118 patients (11%). An exercise test was carried out in less than half of the population and exercise echocardiography in approximately one fifth (Table 2).
CMR was performed in almost half of the cohort. Its incremental value over echocardiography was the assessment of fibrosis (59%), diagnosis in false-negative echocardiograms (6%) and detection of massive LVH (4%).
Risk stratification for sudden cardiac death at baseline (at the time of the first visit)Based on the American Heart Association model for SCD2,20 (Supplementary Table 1), half of the patients had no risk factors, one third had one risk factor and 15% more than one risk factor. Our data also showed that according to the European Society of Cardiology SCD risk score,1,21 the majority of patients had a five-year risk lower than 4%.
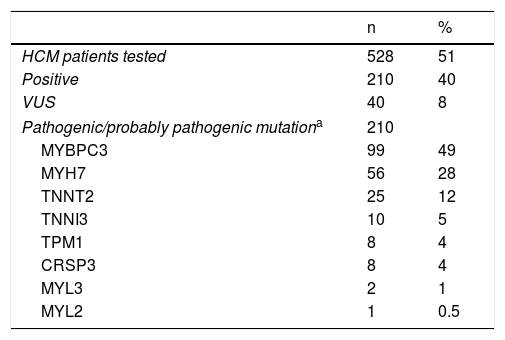
Genetic testingIn total, 51% of the patients had undergone genetic testing and in 40% of these a pathogenic/probably pathogenic mutation was found (Table 3). In this group, when the causative gene mutation was reported, the two most frequent genes were MYBPC3 and MYH7.
Results of genetic testing.15,16
| n | % | |
|---|---|---|
| HCM patients tested | 528 | 51 |
| Positive | 210 | 40 |
| VUS | 40 | 8 |
| Pathogenic/probably pathogenic mutationa | 210 | |
| MYBPC3 | 99 | 49 |
| MYH7 | 56 | 28 |
| TNNT2 | 25 | 12 |
| TNNI3 | 10 | 5 |
| TPM1 | 8 | 4 |
| CRSP3 | 8 | 4 |
| MYL3 | 2 | 1 |
| MYL2 | 1 | 0.5 |
CRSP3: muscle LIM protein; HCM: hypertrophic cardiomyopathy; MYBPC3: cardiac myosin-binding protein C; MYH7: myosin heavy chain; MYL2: regulatory myosin light chain; MYL3: essential myosin light chain; TNNI3: cardiac troponin I; TNNT2: cardiac troponin T; TPM1: tropomyosin; VUS: variants of unknown significance.
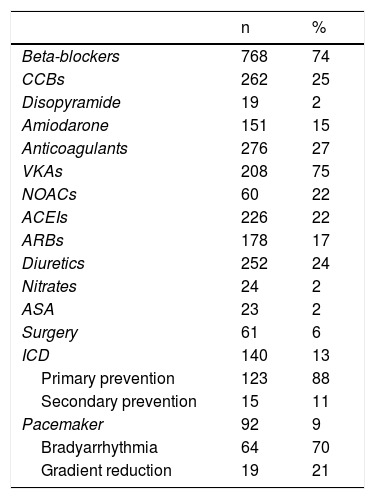
Most patients (n=909; 87%) received medical treatment (Table 4). Septal reduction therapy was performed in 8% of the cohort, 23% of the obstructive group. Cardiac surgery was performed 2.6 times more frequently than ASA. Surgery was performed in 11 centers (of these, only two performed more than 10 surgeries). ASA was performed in four centers (only one reached 10 procedures).
Treatment in the PRo-HCM registry.
| n | % | |
|---|---|---|
| Beta-blockers | 768 | 74 |
| CCBs | 262 | 25 |
| Disopyramide | 19 | 2 |
| Amiodarone | 151 | 15 |
| Anticoagulants | 276 | 27 |
| VKAs | 208 | 75 |
| NOACs | 60 | 22 |
| ACEIs | 226 | 22 |
| ARBs | 178 | 17 |
| Diuretics | 252 | 24 |
| Nitrates | 24 | 2 |
| ASA | 23 | 2 |
| Surgery | 61 | 6 |
| ICD | 140 | 13 |
| Primary prevention | 123 | 88 |
| Secondary prevention | 15 | 11 |
| Pacemaker | 92 | 9 |
| Bradyarrhythmia | 64 | 70 |
| Gradient reduction | 19 | 21 |
ACEIs: angiotensin-converting enzyme inhibitors; ARBs: angiotensin receptor blockers; ASA: alcohol septal ablation; CCBs: calcium channel blockers; ICD: implantable cardioverter defibrillator: NOACs: new oral anticoagulants; VKAs: vitamin K antagonists.
An ICD was implanted in 13% of the population, mainly for primary prevention. A pacemaker was implanted in 9%, usually for conduction disorders.
Follow-up, morbidity and mortalityMean follow-up was 5.3±6.1 years, median 3.3 years (IQR [P25-P75] 1.3-6.5 years). At last assessment, most patients were symptomatic (Figure 2), usually with mild to moderate symptoms. A small number (n=42, 4%) developed systolic dysfunction.
, and the majority had mild to moderate symptoms (right).")
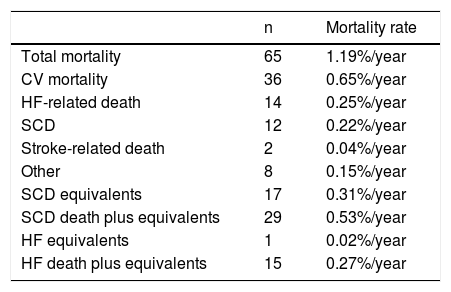
All-cause mortality was 6.2% (Table 5). Cardiovascular mortality was 3.4%, most frequently due to HF, followed by SCD and by stroke-related death.
Mortality in the PRo-HCM registry.
| n | Mortality rate | |
|---|---|---|
| Total mortality | 65 | 1.19%/year |
| CV mortality | 36 | 0.65%/year |
| HF-related death | 14 | 0.25%/year |
| SCD | 12 | 0.22%/year |
| Stroke-related death | 2 | 0.04%/year |
| Other | 8 | 0.15%/year |
| SCD equivalents | 17 | 0.31%/year |
| SCD death plus equivalents | 29 | 0.53%/year |
| HF equivalents | 1 | 0.02%/year |
| HF death plus equivalents | 15 | 0.27%/year |
CV: cardiovascular; HF: heart failure; SCD: sudden cardiac death.
In univariate analysis, 16 of the predefined variables were significantly related to mortality. Multivariate analysis showed four major risk indicators of cardiovascular mortality: late diagnosis (>60 years), family history of SCD, progressive systolic dysfunction and obstructive HCM (Supplementary Table 2).
Of the 12 patients with SCD, seven were between 40 and 65 years old, three were older than 65, and only two were aged under 40 years. In a number of patients SCD was aborted by appropriate ICD shocks in the setting of primary prevention or documented successful in- and/or out-of-hospital resuscitation. Therefore, actual plus aborted SCD occurred in 29 patients.
The incidence of all-cause and cardiovascular mortality was 1.19%/year and 0.65%/year, respectively; the incidence of HF-related death was higher than that of SCD, and the latter was higher than that of stroke. However, the incidence of SCD death plus equivalents was higher than the incidence of HF death, with or without equivalents (Table 5 and Figure 3).
 SCD: sudden cardiac death mortality;
SCD: sudden cardiac death mortality; Kaplan-Meier estimates of the cumulative hazard function for mortality during follow-up. Left: cumulative hazard function for mortality; right: cumulative hazard function for mortality, including sudden cardiac death and heart failure equivalents. See text for explanation. CV: cardiovascular mortality; HF: heart failure mortality; HF+Equiv: heart failure mortality+ equivalents; Stroke: stroke related mortality; SCD: sudden cardiac death mortality; SCD+Equiv: sudden cardiac death mortality + equivalents.
Thromboembolic events occurred in 65 patients (6%) (stroke n=52, TIA n=11, peripheral embolism n=2). Of these, half had documented AF.
Compared with low-volume centers (<15 patients included, n=16), high-volume (>100 patients, n=3) centers had younger patients and more familial HCM and performed more genetic testing, family screening and exclusion of phenocopies (Supplementary Table 3). Additionally, despite the higher number of diagnostic tests and of drug prescriptions of high-volume centers, no major differences in outcomes were found.
DiscussionThe PRo-HCM registry provides a detailed contemporary assessment of the clinical profile, management strategies and outcomes of HCM in Portugal. While most data are consistent with the existing literature,17,18,22 the present findings show elements of novelty and some differences from the guidelines.1,2 Our results are important at both national and international level, as several countries, worldwide, may face similar conditions in the management of the disease.
Epidemiological and sociodemographic dataThe total number of patients included represents about 5% of the estimated prevalence in Portugal,5,7 but up to one third of the Portuguese population with ‘clinical’ HCM.6 Accordingly, this is, to our knowledge, the most comprehensive national HCM registry published.17,18,22 This national effort provides credibility to our data as representative of the real Portuguese scenario. The distribution of patients between referral and community-based centers (four centers included half of the patients and 25 centers the other half) shows that a significant number of patients are followed in non-referral centers. Of note, however, was the low proportion of reported familial HCM, probably reflecting a low rate of systematic family screening programs and/or a referral center bias in another registry.22
Baseline assessmentOver a decade since the publication of another national registry,17 the clinical spectrum of HCM appears very similar, suggesting that its clinical profile is not undergoing major changes in the Western world. The major difference is the older age at diagnosis, with more than one fourth of patients diagnosed over the age of 65 years. This finding may reflect delayed disease penetrance, lack of systematic family screening, and – potentially – an increased diagnostic yield in older patients.1,2 By contrast, the association found in our cohort of a low rate of familial HCM, later age of presentation, and low risk profile, may more closely mirror the real-world disease scenario, reflecting the inclusion of these unselected lower-risk HCM patients in the cohort. Recent reports have in fact identified a lower-risk cohort of HCM patients, with later onset and lower rate of familial disease,23,24 which may explain our findings.
The proportion of obstructive forms in our cohort, about one third, basically reflects patients with obstruction at rest, and is consistent with the existing literature for resting obstruction.1,2 Accordingly, due to the low rate of exercise echocardiography performed,25 many patients with labile obstruction were probably not detected and were classified as non-obstructive, which at first sight suggests a deviation from the guidelines. However, as the recommendations1,2 for the use of exercise echocardiography in non-obstructive HCM at rest are relatively recent, some of these patients, assessed earlier, have not undergone exercise echocardiography and were diagnosed as non-obstructive in this observational study.
Diagnostic tests and sudden cardiac death risk stratification at baselineOur data show a relatively limited penetration of CMR, despite the evidence of its incremental value.1–4 These results reflect its high costs, limited availability, and relatively recent introduction in clinical practice.1–4 By contrast, despite the factors that limit the dissemination of genetic testing1,2 (price, lack of co-payment, low availability), half of the patients underwent genetic study, which in many cases is already part of routine practice.26 The proportion of tests in which a variant was found15,16,27,28 and the relative prevalence of the disease-causing genes is mostly similar to what has been described.1,2,15,16,27,28 However, according to the results provided by the investigators, an unexpectedly high prevalence of pathogenic/probably pathogenic mutations15,16,27,28 was found in the TPM1 and CSRP3 genes.17 These results should be interpreted with caution, because they are derived from CRF raw data that were not centrally reviewed or corrected by the PRo-HCM coordinators.
Both of the contemporary models for SCD risk1,2,20,21 show that our cohort was, at baseline, a low-risk population for SCD, which partially explains the low rate of SCD and of ICD implantations.
TreatmentInvasive septal reduction was offered to almost one fourth of obstructive patients, including those who were mildly symptomatic. Although it cannot be excluded that this rate is biased by the low number of patients detected with labile obstruction, it probably also results from knowledge of the adverse long-term effects of obstruction, as well as from the safety of invasive procedures, which may impact future HCM guidelines.
Of note, the number of surgical myectomies was much higher than the number of ASA procedures, which is partially explained by the late introduction of the latter in Portugal (2009).29 The fact that the two procedures were performed in different centers deserves reflection, taking into account the known effect of expertise on results.1,2
Finally, fewer than 15% of patients received an ICD during follow-up, reflecting the low risk profile of our non-selected population.
Follow-up, morbidity and mortalityOverall, our data suggest that in Portugal, in the era of better diagnostic and therapeutic techniques, HCM has low mortality but high morbidity.
Additionally, despite greater use of diagnostic tests and differences in medical treatment, outcomes of high-volume centers are similar to those of low-volume centers, calling into question the value of HCM centers and of the ‘hub and spoke’ model.7
Outcome data show that the SCD rate in HCM patients in Portugal is very low. Even though this finding may be partially explained by lives saved by successful resuscitation and ICD implantation, the incidence of SCD is still low after including these SCD equivalents in the SCD rate. As a consequence of the efficacy of these preventive measures, HF has become the leading cause of death in HCM patients in Portugal.
Our figures are in overall agreement with those from other groups,30 showing that overall mortality in treated HCM in Portugal is 1.19%, similar to that of the general Portuguese population (around 1.1% year).31 Importantly, at follow-up most patients were symptomatic, confirming that disease morbidity represents a significant burden to patients, health care services and providers. Accordingly, the “contemporary treatable disease”30 has became, at least in Portugal, a “contemporary chronic treatable disease” in which, side by side with ICDs, the role of chronic medical treatment is increasing.
LimitationsDespite their inherent limitations, registries provide realistic geographical data on disease course and management.
The inclusion of mostly symptomatic patients with advanced, established disease (mainly included by HCM referral centers) is a limitation of this registry, providing a biased view of the disease (selection bias, a common limitation of many HCM studies).
Additionally, disease-related mortality is underestimated, as patients who died before diagnosis were not included. This survival bias partially explains the low rate of events, especially the low rate of SCD.
Children were excluded because of important clinical differences.1,2
Future directionsThe identification, at a national level, of discrepancies between our data and the guidelines is an important finding, warranting a national effort to correct them (for instance to include exercise echocardiography as a standard initial assessment of non-obstructive HCM at rest, to better detect labile obstruction).
Because of the large volume of data, we were unable to cover some important topics in depth. Accordingly, further work will be directed at comparisons between subgroups, addressing family screening, genetic testing (including founder effects, differences in phenotype between genes, and analysis of specific mutations considered as pathogenic/probably pathogenic by the investigators), awareness of phenocopies (for instance Fabry disease), and detailed assessment of clinical HCM profiles.
ConclusionsThe PRo-HCM registry provides comprehensive data on the management of HCM in Portugal in the era of genetics, CMR, ICDs and ASA, and indicates the need for better access to resources and some deviations from guidelines.
Contemporary HCM in Portugal is characterized by relatively advanced age at diagnosis, and a high proportion of invasive treatment of obstructive forms at rest. Long-term mortality is low, and HF is the most common cause of death followed by SCD (excluding equivalents). However, morbidity remains considerable, emphasizing the need for disease-specific treatments that impact the natural history of the disease.
Funding (unrestricted grants to the Portuguese Society of Cardiology for all myocardial and pericardial disease registries)Gold: Jaba Recordati, Merck Serono, Sanofi-Genzyme, Shire; Silver: Medinfar, Servier
IOi was supported by the Italian Ministry of Health (“LVH in aortic valve disease and HCM: genetic basis, biophysical correlates and viral therapy models” (RF-2013-02356787), and NET-2011-02347173 (“Mechanisms and treatment of coronary microvascular dysfunction in patients with genetic or secondary LVH”); and by Telethon Italy (GGP13162).
Conflicts of interestThe authors have no conflicts of interest to declare.
We are grateful to CNCDC staff, especially Dr. Sandra Corker, and to Infortucano staff.
Centro Hospitalar de Leiria: Joana Correia; Centro Hospitalar de Lisboa Norte - Hospital de Santa Maria: Dulce Brito; Centro Hospitalar de Lisboa Ocidental, Serviço de Cardiologia: João Abecasis; Centro Hospitalar de Lisboa Ocidental - Hospital São Francisco Xavier - Serviço de Medicina III: Cândida Fonseca; Centro Hospitalar de Trás os Montes e Alto Douro - Hospital São Pedro: Carla Alexandra R. Araújo; Centro Hospitalar de Vila Nova de Gaia/Espinho: Conceição Fonseca; Centro Hospitalar do Algarve - Hospital de Faro: Nuno Marques; Centro Hospitalar do Alto Ave - Hospital da Senhora da Oliveira: Olga Azevedo; Centro Hospitalar do Baixo Vouga - Hospital Infante D. Pedro: José António Nobre dos Santos; Centro Hospitalar do Oeste Norte - Centro Hospitalar das Caldas da Rainha: Ana Filipa Pereira Rodrigues; Centro Hospitalar do Porto - Hospital de Santo António: Patrícia Fernandes Rodrigues; Centro Hospitalar do Tâmega e Sousa - Unidade Padre Américo: Maria Conceição Queirós; Centro Hospitalar e Universitário de Coimbra - Cardiologia B - Hospital Geral: Joana Delgado Silva; Centro Hospitalar Tondela Viseu - Hospital de São Teotónio: Carlos Emanuel Correia; CUF Infante Santo Hospital: Pedro Matos; Hospital Beatriz Ângelo: Luís Sargento; Hospital da Luz Lisboa: Nuno Cardim; Hospital das Forças Armadas: Sara Ferreira; Hospital de Braga: Nuno Salomé: Hospital de Santa Maria Maior de Barcelos - Serviço Cardiologia: Alexandra Sousa; Hospital de Santo Espírito de Angra do Heroísmo: Rute Couto; Hospital de São João: Elisabete Martins; Hospital do Espírito Santo: Agostinho Caeiro; Hospital Garcia de Orta: Luís Rocha Lopes; Hospital Prof. Doutor Fernando Fonseca: Francisco Madeira; Hospital SAMS: Berta Carola; HPP Hospital de Cascais - Hospital Dr. José de Almeida: Gonçalo Proença; Unidade Local de Saúde da Guarda - Hospital Sousa Martins: Maria Cristina Gamboa.
