

Cardiomyopathies are rare diseases of the heart muscle, of multiple causes, that manifest with various structural and functional phenotypes but are invariably associated with cardiac dysfunction. Dilated cardiomyopathy is the commonest cardiomyopathy in children, and the majority present before one year of age. Its etiology may be acquired or genetic. Myocarditis is an important cause and is responsible for the majority of acquired cases. Inherited (familial) forms of dilated cardiomyopathy may occur in 25-50% of patients. Echocardiographic and tissue Doppler studies are the basis for diagnosis of dilated cardiomyopathy in most patients. Marked dilatation of the left ventricle with global hypokinesis is the hallmark of the disease. This review will cover the classification, epidemiology and management of newborns with dilated cardiomyopathy. In particular, a comprehensive and up-to-date review of the genetic study of dilated cardiomyopathy and of detailed echocardiographic assessment of these patients will be presented.
As cardiomiopatias são doenças raras do músculo cardíaco, de múltiplas causas, que se manifestam com vários fenótipos estruturais e funcionais, mas invariavelmente associadas a disfunção cardíaca. A cardiomiopatia dilatada é a forma mais comum em crianças, apresentando-se maioritariamente antes do ano de idade. A sua etiologia pode ser adquirida ou genética. A miocardite é uma importante causa, responsável pela maioria dos casos adquiridos. As formas hereditárias (familiares) de cardiomiopatia dilatada podem ocorrer em 25-50% dos pacientes. Os estudos ecocardiográficos e Doppler são a base para o seu diagnóstico, caracterizando-se por uma acentuada dilatação do ventrículo esquerdo com hipocinesia global. Neste artigo serão abordadas a classificação, a epidemiologia e a abordagem dos recém-nascidos com cardiomiopatia dilatada. Em particular, será apresentada uma revisão abrangente e atualizada da avaliação genética desta entidade, bem como aspetos detalhados da ecocardiografia nestes pacientes.
angiotensin-converting enzyme
autosomal dominant
American Heart Association
autosomal recessive
arrhythmogenic right ventricular cardiomyopathy/dysplasia
American Society of Echocardiography
B-type natriuretic peptide
cardiomyopathy
cardiovascular magnetic resonance
cardiac output
dilated cardiomyopathy
diastolic dysfunction
ejection fraction
European Society of Cardiology
hypertrophic cardiomyopathy
heart failure
inborn errors of metabolism
International Society and Federation of Cardiology
left ventricular
left ventricular ejection fraction
mitral regurgitation
restrictive cardiomyopathy
World Health Organization
Cardiomyopathies (CMs) are a group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation and are due to a variety of causes that frequently are genetic. CMs are either confined to the heart or are a part of generalized systemic disorders, often leading to cardiovascular death or progressive heart failure (HF)-related disability.1
Different definitions and nomenclature have been proposed for this important and heterogeneous group of diseases. Neonatal CMs are diseases of the heart muscle of the neonate in which the myocardium is affected without primary abnormalities of the valves, great vessels or septum.1 They include a variety of myocardial disorders that manifest with various structural and functional phenotypes. Neonatal CMs account for about 1% of childhood cardiac disease, with an estimated incidence of 10:100000 live births, and are responsible for 10% of all pediatric cardiac deaths.2
In this review we focus on clinical aspects, management, prognosis and follow-up in dilated cardiomyopathy (DCM) of the newborn.
Definition and classificationCMs are rare diseases of the heart muscle, of multiple causes, that manifest with various structural and functional phenotypes but are invariably associated with cardiac dysfunction.
At present there is no consensus on how to classify CMs on the basis of etiology, physiology or treatment, as their origin and pathophysiology are not well understood.
The first classification to take anatomical presentation into consideration was the 1995 World Health Organization (WHO)/International Society and Federation of Cardiology (ISFC) report, which divided myocardial disease into DCM, hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy (RCM), arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D), and unclassified cardiomyopathies.3
The American Heart Association (AHA) and the European Society of Cardiology (ESC) published classifications based on etiology and pathophysiology.4,5 In the AHA classification, CMs are categorized into two groups: primary (predominantly involving the heart) and secondary (accompanied by systemic involvement of other organs). Primary CMs are subdivided into genetic, mixed (predominantly non-genetic; less commonly genetic), or acquired. Genetic CMs include hypertrophic cardiomyopathy, ARVC/D, left ventricular noncompaction, glycogen storage diseases, conduction defects, mitochondrial myopathies, and ion channel disorders. Mixed CMs include DCM and RCM. Acquired CMs include myocarditis, stress-induced (Takotsubo), peripartum and tachycardia-induced CM, and CM in infants of insulin-dependent diabetic mothers.6
The AHA and ESC classification systems differ from the earlier WHO/ISFC classification in emphasizing the distinction between familial/genetic and non-familial/non-genetic causes of CM and excluding heart disease. A more recent phenotype-genotype-based classification, endorsed by the World Heart Federation (formerly ISFC), was published in 2013. This proposed system, inspired by the TNM staging of malignant tumors, does not include ion channelopathies. It addresses five attributes, abbreviated as MOGE(S): (1) the morphofunctional (M) notation provides a descriptive phenotypic diagnosis (e.g., MD=dilated cardiomyopathy); (2) the organ involvement (O) notation indicates whether heart and/or extracardiac involvement is present (e.g., OH+K=heart and kidney involvement); (3) the genetic or familial inheritance (G) notation indicates the nature of genetic transmission (e.g., GAD=autosomal dominant); (4) the etiological annotation (E) provides a description of the specific cause (e.g., the specific gene and mutation as in EG-MYH7[p.Arg403Glu]); (5) an additional functional status (S) term is optional (e.g., SC-II=stage C disease in New York Heart Association functional class II).7,8
EpidemiologyDCM is the commonest cardiomyopathy in children, accounting for up to three-fifths of cases.6 In the USA, the reported incidence of DCM is 0.57 cases per 100000 children per year,9 while in Finland it is 2.6 per 100000 children10 and 0.87 cases per 100000 individuals older than 16 years in the UK.11 DCM is reportedly more common in boys than in girls, and some forms are clearly X-linked. One large longitudinal population-based study reported an incidence in boys and girls of 0.66 and 0.47 cases per 100000 per year, respectively.12 All age-groups are affected. However, studies suggest that the majority of children with DCM present before one year of age.6,9 The same population-based study reported an incidence in infants (<1 year) and older children of 4.40 and 0.34 cases per 100000 per year, respectively.12 Fetal presentation is uncommon.9
EtiologyDCM may be a consequence of a variety of cardiac and systemic pathologies and therefore differential diagnosis is quite broad. In most patients no identifiable cause is found,13–15 with some studies reporting identification of the cause of DCM in only 34% of patients.12–14
Acquired conditions associated with DCM in newborns and infants are listed in Table 1.6,16–19 Myocarditis is responsible for the majority of cases.13,20 Viral infection is the most common cause of myocarditis and has been implicated in the development of DCM.21 The initial immune response limits the degree of viremia early during infection and protects against myocarditis. If, however, this response is insufficient, the virus may not be eliminated and myocyte injury may ensue via one of two mechanisms: direct cytotoxicity via receptor-mediated entry of the virus into cardiac myocytes, or an adverse autoimmune response induced by persisting viral genomic fragments.21
Acquired conditions associated with dilated cardiomyopathy.
| Infectious diseases |
| Viral |
| Adenovirus, coxsackie, cytomegalovirus, human immunodeficiency virus, influenza, varicella, hepatitis C, echovirus, parvovirus B19, Epstein-Barr, human herpesvirus 6, others |
| Bacterial |
| Streptococcus spp., Treponema pallidum, Mycoplasma pneumoniae, Mycobacterium, Leptospira, others |
| Spirochetal |
| Lyme disease |
| Fungal |
| Candida spp., Aspergillus, Coccidioides, Cryptococcus, Histoplasma, others |
| Parasitic |
| Toxoplasmosis, trypanosomiasis, others |
| Arrhythmias |
| Supraventricular tachycardia (atrial flutter, ectopic atrial tachycardia), ventricular tachycardia, bradycardia |
| Ischemia |
| Birth asphyxia, hypoxia, coronary artery malformation |
| Toxic |
| Antipsychotic/antidepressant drugs, cocaine, ethanol, chemotherapeutic agents, antiretroviral drugs, phenothiazines, chloroquine, clozapine, anthracycline, amphetamines, sulfonamide, penicillin, cephalosporins, diuretics, dobutamine, interleukin-2, copper, lead, lithium, cobalt, mercury, carbon monoxide, beryllium, radiation |
| Endocrine |
| Hyper/hypothyroidism, excess catecholamines (pheochromocytoma, neuroblastoma), congenital adrenal hyperplasia, diabetes, growth hormone excess or deficiency, Cushing's syndrome, hyperparathyroidism |
| Metabolic |
| Disorders of glycogen metabolism, disorders of lipid metabolism, defects of beta-oxidation enzymes, carnitine deficiency syndromes, fatty acid transport defects |
| Nutritional deficiencies |
| Selenium, thiamine, vitamin E, phosphate |
| Electrolyte and renal abnormalities |
| Hypocalcemia, hypophosphatemia, uremia |
| Deposition diseases |
| Hemochromatosis, amyloidosis |
| Autoimmune disorders |
| Systemic lupus erythematosus, dermatomyositis, scleroderma, rheumatoid arthritis, sarcoidosis, giant cell arteritis, inflammatory bowel disease, Churg-Strauss syndrome, Wegener's granulomatosis, Takayasu arteritis, Kawasaki disease, polyarteritis nodosa, celiac disease |
| Systemic disorders |
| Osteogenesis imperfecta, leukemia |
| Miscellaneous |
| Heat stroke, hypothermia |
Adapted from6,16–19.
According to the Pediatric Cardiomyopathy Registry, as well as other authors, inborn errors of metabolism (IEM) account for 6.8% of cases of dilated cardiomyopathy; however, their data demonstrate that the most common adaptive response of the heart in IEM is hypertrophy, with or without dilatation. The same series reported myocarditis accounting for more than 50% of the known cause of dilated cardiomyopathy, consistent with the above data.15 In a North American population-based study, among patients with IEM, the largest subgroup was those with mitochondrial disorders, responsible for almost half of the cases.12
Genetic (familial) forms of DCM occur in 25%-50% of patients, although this prevalence may be underestimated (see below).13,21
Neuromuscular diseases are an important cause of DCM, being responsible for nearly a quarter of cases with an identified cause in some series. Among patients with neuromuscular disease and DCM, about 80% have Duchenne muscular dystrophy.12
In cases in which an underlying pathology or a first-degree family relative cannot be identified, the patient is diagnosed with idiopathic DCM.13,21
GeneticsFamilial disease occurs in over a third of adult patients, although the reported prevalence in children is much lower (one-twentieth to one-sixth).22 This is probably an underestimate resulting from lack of awareness of the inherited nature of the condition among pediatricians, and possibly a higher prevalence of metabolic and syndromic causes in children.22
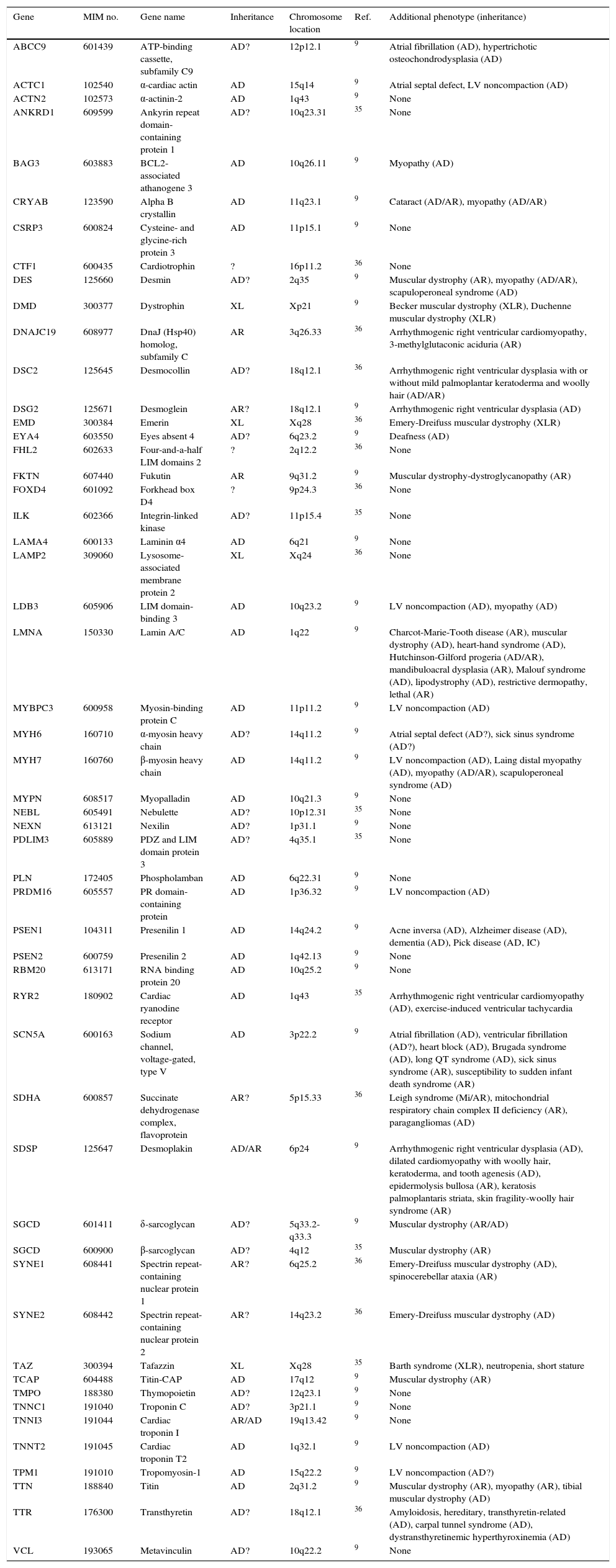
Modes of inheritance include autosomal dominant (AD) with incomplete penetrance due to modifier genes and environmental factors, autosomal recessive (AR), X-linked and mitochondrial.13 More than 40 genes have been identified as harboring mutations that cause DCM.21 Genetic testing has advanced from basic scientific research to clinical application and has been incorporated as part of patient assessment for suspected inherited CMs. Genes in the OMIM® (Online Mendelian Inheritance in Man) catalog20 involved in familial DCM, as well as other genes suspected of being involved in DCM, are shown in Table 2.20–37
Genes involved in familial dilated cardiomyopathy.
| Gene | MIM no. | Gene name | Inheritance | Chromosome location | Ref. | Additional phenotype (inheritance) |
|---|---|---|---|---|---|---|
| ABCC9 | 601439 | ATP-binding cassette, subfamily C9 | AD? | 12p12.1 | 9 | Atrial fibrillation (AD), hypertrichotic osteochondrodysplasia (AD) |
| ACTC1 | 102540 | α-cardiac actin | AD | 15q14 | 9 | Atrial septal defect, LV noncompaction (AD) |
| ACTN2 | 102573 | α-actinin-2 | AD | 1q43 | 9 | None |
| ANKRD1 | 609599 | Ankyrin repeat domain-containing protein 1 | AD? | 10q23.31 | 35 | None |
| BAG3 | 603883 | BCL2-associated athanogene 3 | AD | 10q26.11 | 9 | Myopathy (AD) |
| CRYAB | 123590 | Alpha B crystallin | AD | 11q23.1 | 9 | Cataract (AD/AR), myopathy (AD/AR) |
| CSRP3 | 600824 | Cysteine- and glycine-rich protein 3 | AD | 11p15.1 | 9 | None |
| CTF1 | 600435 | Cardiotrophin | ? | 16p11.2 | 36 | None |
| DES | 125660 | Desmin | AD? | 2q35 | 9 | Muscular dystrophy (AR), myopathy (AD/AR), scapuloperoneal syndrome (AD) |
| DMD | 300377 | Dystrophin | XL | Xp21 | 9 | Becker muscular dystrophy (XLR), Duchenne muscular dystrophy (XLR) |
| DNAJC19 | 608977 | DnaJ (Hsp40) homolog, subfamily C | AR | 3q26.33 | 36 | Arrhythmogenic right ventricular cardiomyopathy, 3-methylglutaconic aciduria (AR) |
| DSC2 | 125645 | Desmocollin | AD? | 18q12.1 | 36 | Arrhythmogenic right ventricular dysplasia with or without mild palmoplantar keratoderma and woolly hair (AD/AR) |
| DSG2 | 125671 | Desmoglein | AR? | 18q12.1 | 9 | Arrhythmogenic right ventricular dysplasia (AD) |
| EMD | 300384 | Emerin | XL | Xq28 | 36 | Emery-Dreifuss muscular dystrophy (XLR) |
| EYA4 | 603550 | Eyes absent 4 | AD? | 6q23.2 | 9 | Deafness (AD) |
| FHL2 | 602633 | Four-and-a-half LIM domains 2 | ? | 2q12.2 | 36 | None |
| FKTN | 607440 | Fukutin | AR | 9q31.2 | 9 | Muscular dystrophy-dystroglycanopathy (AR) |
| FOXD4 | 601092 | Forkhead box D4 | ? | 9p24.3 | 36 | None |
| ILK | 602366 | Integrin-linked kinase | AD? | 11p15.4 | 35 | None |
| LAMA4 | 600133 | Laminin α4 | AD | 6q21 | 9 | None |
| LAMP2 | 309060 | Lysosome-associated membrane protein 2 | XL | Xq24 | 36 | None |
| LDB3 | 605906 | LIM domain-binding 3 | AD | 10q23.2 | 9 | LV noncompaction (AD), myopathy (AD) |
| LMNA | 150330 | Lamin A/C | AD | 1q22 | 9 | Charcot-Marie-Tooth disease (AR), muscular dystrophy (AD), heart-hand syndrome (AD), Hutchinson-Gilford progeria (AD/AR), mandibuloacral dysplasia (AR), Malouf syndrome (AD), lipodystrophy (AD), restrictive dermopathy, lethal (AR) |
| MYBPC3 | 600958 | Myosin-binding protein C | AD | 11p11.2 | 9 | LV noncompaction (AD) |
| MYH6 | 160710 | α-myosin heavy chain | AD? | 14q11.2 | 9 | Atrial septal defect (AD?), sick sinus syndrome (AD?) |
| MYH7 | 160760 | β-myosin heavy chain | AD | 14q11.2 | 9 | LV noncompaction (AD), Laing distal myopathy (AD), myopathy (AD/AR), scapuloperoneal syndrome (AD) |
| MYPN | 608517 | Myopalladin | AD | 10q21.3 | 9 | None |
| NEBL | 605491 | Nebulette | AD? | 10p12.31 | 35 | None |
| NEXN | 613121 | Nexilin | AD? | 1p31.1 | 9 | None |
| PDLIM3 | 605889 | PDZ and LIM domain protein 3 | AD? | 4q35.1 | 35 | None |
| PLN | 172405 | Phospholamban | AD | 6q22.31 | 9 | None |
| PRDM16 | 605557 | PR domain-containing protein | AD | 1p36.32 | 9 | LV noncompaction (AD) |
| PSEN1 | 104311 | Presenilin 1 | AD | 14q24.2 | 9 | Acne inversa (AD), Alzheimer disease (AD), dementia (AD), Pick disease (AD, IC) |
| PSEN2 | 600759 | Presenilin 2 | AD | 1q42.13 | 9 | None |
| RBM20 | 613171 | RNA binding protein 20 | AD | 10q25.2 | 9 | None |
| RYR2 | 180902 | Cardiac ryanodine receptor | AD | 1q43 | 35 | Arrhythmogenic right ventricular cardiomyopathy (AD), exercise-induced ventricular tachycardia |
| SCN5A | 600163 | Sodium channel, voltage-gated, type V | AD | 3p22.2 | 9 | Atrial fibrillation (AD), ventricular fibrillation (AD?), heart block (AD), Brugada syndrome (AD), long QT syndrome (AD), sick sinus syndrome (AR), susceptibility to sudden infant death syndrome (AR) |
| SDHA | 600857 | Succinate dehydrogenase complex, flavoprotein | AR? | 5p15.33 | 36 | Leigh syndrome (Mi/AR), mitochondrial respiratory chain complex II deficiency (AR), paragangliomas (AD) |
| SDSP | 125647 | Desmoplakin | AD/AR | 6p24 | 9 | Arrhythmogenic right ventricular dysplasia (AD), dilated cardiomyopathy with woolly hair, keratoderma, and tooth agenesis (AD), epidermolysis bullosa (AR), keratosis palmoplantaris striata, skin fragility-woolly hair syndrome (AR) |
| SGCD | 601411 | δ-sarcoglycan | AD? | 5q33.2-q33.3 | 9 | Muscular dystrophy (AR/AD) |
| SGCD | 600900 | β-sarcoglycan | AD? | 4q12 | 35 | Muscular dystrophy (AR) |
| SYNE1 | 608441 | Spectrin repeat-containing nuclear protein 1 | AR? | 6q25.2 | 36 | Emery-Dreifuss muscular dystrophy (AD), spinocerebellar ataxia (AR) |
| SYNE2 | 608442 | Spectrin repeat-containing nuclear protein 2 | AR? | 14q23.2 | 36 | Emery-Dreifuss muscular dystrophy (AD) |
| TAZ | 300394 | Tafazzin | XL | Xq28 | 35 | Barth syndrome (XLR), neutropenia, short stature |
| TCAP | 604488 | Titin-CAP | AD | 17q12 | 9 | Muscular dystrophy (AR) |
| TMPO | 188380 | Thymopoietin | AD? | 12q23.1 | 9 | None |
| TNNC1 | 191040 | Troponin C | AD? | 3p21.1 | 9 | None |
| TNNI3 | 191044 | Cardiac troponin I | AR/AD | 19q13.42 | 9 | None |
| TNNT2 | 191045 | Cardiac troponin T2 | AD | 1q32.1 | 9 | LV noncompaction (AD) |
| TPM1 | 191010 | Tropomyosin-1 | AD | 15q22.2 | 9 | LV noncompaction (AD?) |
| TTN | 188840 | Titin | AD | 2q31.2 | 9 | Muscular dystrophy (AR), myopathy (AR), tibial muscular dystrophy (AD) |
| TTR | 176300 | Transthyretin | AD? | 18q12.1 | 36 | Amyloidosis, hereditary, transthyretin-related (AD), carpal tunnel syndrome (AD), dystransthyretinemic hyperthyroxinemia (AD) |
| VCL | 193065 | Metavinculin | AD? | 10q22.2 | 9 | None |
AD: autosomal dominant; AR: autosomal recessive; IC: isolated case; LV: left ventricular; Mi: mitochondrial; Ref.: reference; XL: X-linked; XLR: X-linked recessive.
Idiopathic DCM is estimated to be associated with a familial component in 20-48% of cases, with AD inheritance being the predominant pattern of transmission, while X-linked, AR, and mitochondrial inheritance are less common.20–36 Two major forms of AD DCM have been recognized: isolated (or pure) DCM, and DCM associated with cardiac conduction system disease. Its pathophysiology is poorly understood. Some mutations result in DCM and conduction disease alone,20 while others lead to juvenile-onset muscular dystrophies, including Emery-Dreifuss muscular dystrophy and familial partial lipodystrophy with insulin-resistant diabetes.20
X-linked inheritance accounts for about 5% of familial cases of DCM,14,20 and may occur in Duchenne, Becker, and Emery-Dreifuss muscular dystrophies.22 Mutations in mitochondrial tRNAs, which have matrilineal inheritance, affect cardiac function and hearing. They are usually associated with encephalopathy, skeletal myopathies and metabolic abnormalities.20
Clinical presentationDCM affects both genders almost equally,38–40 with a slight male predominance being described in some reviews,9,12 and in the pediatric population it is most common in the first year of life38–40 with about 20% presenting in the neonatal period.40
Infants with DCM often present with symptoms of HF13,39–42 such as tachypnea, dyspnea, tachycardia, feeding difficulty and failure to thrive.38–41 These symptoms are usually progressive and correlate with the degree of myocardial dysfunction.39 Occasionally presentation may also be with acute pulmonary edema41 or arrhythmias.13,40,42
Physical examination can reveal a displaced apical impulse,13,41 muffled heart sounds41 with a third heart sound,13,41 a mitral regurgitation murmur13,41 or signs of pulmonary congestion.13 Signs of systemic venous congestion including hepatomegaly and peripheral edema may be present13,41 but are less common in infants.41
A thorough physical examination including assessment of dysmorphology and ophthalmologic examination should be performed and can provide important diagnostic clues. Patients with IEM may present with signs of multiple organ dysfunction such as encephalopathy, muscle weakness, hypotonia and recurrent vomiting, and the presence of dysmorphic features may indicate malformation syndromes or storage diseases.41
A careful family history is also essential. It is important to identify any family members who have been diagnosed with DCM or who have a history of a thromboembolic event or sudden cardiac death before the age of 30-35 years.13 Attention should also be paid to consanguinity40 and to a family history of muscular dystrophy and signs or symptoms of inherited metabolic disease.17
Initial workupChest X-rayPatients with DCM usually present an increased cardiothoracic ratio, reflecting enlarged left cardiac chambers. In patients with severely compromised cardiac function, signs of pulmonary congestion (from increased vascular markings to pleural effusion) may be present.
Laboratory testsCardiac biomarkers such as creatinine kinase, troponin I and troponin T may be increased in patients with DCM, particularly in the setting of inflammatory cardiac involvement. B-type natriuretic peptide (BNP) will be elevated if chronic HF is present. Although usually associated with HCM, oxidative phosphorylation defects can present as DCM, particularly in the setting of multiorgan disease, thus warranting screening for lactic acidosis in these patients, who may present with elevated lactate and lactate/pyruvate ratio and decreased free carnitine with a relative increase in acylcarnitine species.43,44 DCM has been described in the setting of transient neonatal hypocalcemia and calcium/magnesium ratio should be obtained in patients presenting with cardiac chamber dilation, particularly if accompanied by seizures.45
ElectrocardiographyAlthough usually showing sinus tachycardia and nonspecific ST-segment and T-wave changes in the inferior and lateral leads, the electrocardiogram in DCM can be normal. Q waves in the septal leads may be present and arrhythmias, supraventricular and ventricular, occur in up to half of the patients. Atrioventricular block may be present, particularly in patients carrying mutations of the lamin A/C gene26,46 and in patients with DCM secondary to congenital complete atrioventricular block and neonatal lupus.47–49
Echocardiographic assessmentEchocardiography and Doppler studies are the basis for the diagnosis of DCM in most patients. Marked left ventricular (LV) dilatation with global hypokinesia is the hallmark of the disease. LV fractional shortening is usually less than 25% and LV ejection fraction (LVEF) is less than 50%. The LV walls are thin and areas of dyskinesia may be observed. The left atrium is also dilated, and the mitral valve leaflets show sluggish movement. The anterior leaflet does not appose to the interventricular septum, giving an increased E point septal separation on the M-mode pictures. The M-mode also clearly reveals the limited excursions of the anterior and posterior leaflets during diastole. Doppler studies show varying degrees of mitral regurgitation (MR) secondary to LV dilatation and possible papillary muscle dysfunction. MR is more prominent in follow-up studies after beginning treatment, when cardiac output and systolic function are improved. LV ejection parameters show decreases in peak velocity and peak acceleration, prolongation of the pre-ejection period, and decrease in ejection time. These flow measurements are dependent on loading conditions. Longitudinal function can be assessed by serial measurements of the mitral and tricuspid valve displacements in systole. Speckle tracking strain can complement tissue Doppler imaging to identify dyssynchrony.50 Real-time three-dimensional echocardiography also helps to assess dyssynchrony.51
Long-standing cases show evidence of pulmonary hypertension in the form of right ventricular dilatation and hypertrophy and tricuspid regurgitation. Tricuspid regurgitation and pulmonary regurgitation velocities give an estimate of the pulmonary artery systolic and diastolic pressures, respectively.
In severe cases, swirling echodensity can be observed along the outer ventricular wall, moving from the mitral valve towards the aortic valve. Occasionally thrombi can be visualized in the LV apex and in the left atrium. Pericardial effusion may also be present.
Several studies have reported that the directions of myocardial fibers varies gradually from subendocardium to subepicardium, and the subendocardium is composed of myocardial fibers oriented in different directions from those of the subepicardium.52–55 Rotation of the LV apex relative to the base is related to myocardial contractility and multilayer fiber orientations, and is a key parameter of cardiac performance.56–58 LV torsion and untwisting overall showed age-related increases, and when normalized by LV length, they showed higher values in infancy and middle age.59 Recent studies reported impaired rotation and torsion in certain clinical conditions presenting as DCM.60,61 Jin et al.62 demonstrated that global torsion was decreased in DCM patients. In normal controls, apical rotation was consistently counterclockwise (positive), but basal rotation was clockwise (negative) during systole except early systole. LV rotation was regionally heterogeneous and abnormal in magnitude and patterns in DCM. Apical segmental and global systolic counterclockwise rotation was decreased or abolished. The marked heterogeneity of regional LV function has frequently been noted in patients with non-ischemic DCM. All segmental and global peak torsion was significantly decreased in DCM patients. Loss of LV torsion occurred mainly by the diminution of counterclockwise apical rotation and was augmented by somewhat less reduction in clockwise basal rotation. Additionally they showed that global LV peak circumferential strain and peak radial strain were reduced in the DCM group.62
Possible mechanisms for functional heterogeneity include regional variations in wall stress, myocardial contractile efficiency, oxidative metabolism, myocardial perfusion and interstitial fibrosis.63–66 With depressed, delayed and disorganized untwisting, reflecting ineffective uncoiling of the myocardium, the DCM ventricle fails to generate the effective pressure fall of isovolumic relaxation time and the subsequent suction phase. The mechanisms underlying the changes in torsion dynamics are unknown but probably encompass many factors associated with cardiomyopathic conditions, including LV dilatation, remodeling of cardiomyocytes and the connective tissue matrix, slowed transmural fiber activation, and alterations in excitation-contraction coupling.67–73
Diagnosis of diastolic dysfunction (DD) in children with CM is challenging. Although reference values exist for individual parameters of diastolic function, there is little guidance available to the practicing clinician for diagnosis and grading of DD in children with CM. The pediatric cardiologist must therefore rely on existing guidelines and recommendations derived from adult studies. Echocardiographic assessment is based on the integration of information obtained from mitral inflow patterns, pulmonary venous Doppler and tissue Doppler imaging.74 Echocardiographic studies in adult patients describe a progression of DD along a continuum of increasing severity ranging from normal, through delayed relaxation and pseudonormal filling to a restrictive filling pattern.75–77 The recent guidelines of the American Society of Echocardiography are based on this progression, but do not deal with their application to infants, children or adolescents. In a recent study Dragulescu et al. reported that diastolic parameters are frequently normal in children with overt and even severe cardiac dysfunction. Additionally they showed that there is frequent discordance between peak early diastolic tissue velocity at the mitral annulus (E′) and left atrial volume criteria, hindering the use of the diagnostic flowcharts in the adult guidelines, and also that discordant and overlapping criteria preclude more precise grading of DD in the majority of patients. These and other factors lead to overall poor agreement among observers, even in a research setting.78 Published normal values for diastolic parameters are based on relatively small populations, especially when divided by age, and present a wide range of values.79–81 Criteria consistent with delayed relaxation are distinctly uncommon in children.78 Almost half of DCM patients have diastolic parameters within the normal range but severe systolic dysfunction. Current echocardiographic criteria are inadequate to diagnose and classify DD in pediatric CM. The same researchers78 reported that when DD was diagnosed or suspected, discrepancies between diastolic parameters or diagnostic criteria within individual patients were common. This adversely affects interpretation of diastolic function, the ability to grade DD, and interobserver agreement. These problems are not exclusive to the pediatric population: recent studies in adult populations have demonstrated important disagreements between investigators in classifying pseudonormal and restrictive patterns of DD. Agreement was relatively good for normal and delayed relaxation and was moderate for estimation of filling pressures.82 Among the various echocardiographic parameters of diastolic function E′, mitral E wave deceleration time, and left atrial volume indexed to body surface area are likely to be the most useful in the assessment of diastolic function in children with CM. However, even these parameters were often discrepant in the individual child.80 Early diastolic tissue velocities, particularly medial mitral valve early diastolic tissue velocities, seemed to differentiate patients from controls better than lateral mitral valve early diastolic tissue velocities or mitral valve peak early diastolic inflow velocity-to-mean early diastolic tissue velocities ratio, implying decreased recoil or possibly delayed relaxation.83 Deceleration time also appeared to differentiate patients from controls, albeit with considerable overlap, possibly related in part to pseudonormalization.78
InvestigationThe existence of a first-degree relative with DCM or sudden cardiac death under the age of 30-35 years is suggestive of a familial form of DCM.13
Viral serology and culture should be performed to rule out myocarditis. Titers of neutralizing antibodies to circulating viruses are generally elevated over a period of 2-4 weeks.19 Elevated virus-specific IgM class antibodies are indicative of a recent infection.16 Serum levels of creatinine kinase may provide a clue for a diagnosis of DCM. Elevated troponin I and T levels may suggest an inflammatory or ischemic cause. Plasma BNP levels are elevated in children with chronic HF, and predict rates of survival, hospitalization, and listing for cardiac transplantation.22
A detailed sequential assessment of the patient is mandatory in order to rule out the secondary etiologies reported in Table 1. If neither family history nor an underlying cause is detected, idiopathic DCM is a possible diagnosis.13
Cardiovascular magnetic resonance (CMR) enables noninvasive characterization of the myocardium with high spatial resolution and can detect anomalies of the coronary arteries non-invasively and without the use of radiation. In the acute phase of myocarditis, CMR can be used to assess ventricular function and to identify regional wall motion abnormalities, myocardial inflammatory changes and cell necrosis/fibrosis. In chronic myocarditis, CMR can be used to monitor biventricular function and demonstrate resolution of inflammation and myocardial remodeling.84
Endomyocardial biopsy may be considered in order to rule out conditions with a similar clinical presentation to idiopathic DCM, but which may require different treatment, such as storage diseases.13 Myocardial biopsy material enables inflammatory and noninflammatory states to be distinguished. Biopsies in children are technically more difficult than in adults, but may be crucial to the diagnosis and determination of the etiology.14
Clinical genetic testing for DCM is indicated in patients with familial DCM. Testing is now available in panels of genes, which increase testing sensitivity and also decrease cost. However, while testing greater numbers of genes increases sensitivity, to perhaps as high as 50%, it also increases the likelihood that variants of uncertain significance will complicate interpretation. Clinicians ordering genetic testing should ensure that all tested patients receive pre- and post-test genetic counseling.20
TreatmentThe main goals of treatment include symptom control and prevention of disease progression and complications, including congestive HF, arrhythmias, sudden death and thromboembolic events.6,13,85 Guidelines for drug therapy in neonates are largely based on consensus and extrapolation from data on children and adults.86,87 Therapeutic interventions in patients with DCM depend on several factors, including the degree of ventricular dysfunction and HF, and where the patient is being treated (acutely in an intensive care unit or in a more chronic and controlled setting such as home care.85
Discussion of specific treatments, medical or surgical, related to the underlying disease, falls outside the scope of this article, due the variety of situations involved.
Acute managementThe initial approach to any patient at risk of circulatory collapse should include oxygen, ventilatory support, sedation and vascular access, but discussion of these general measures is beyond the scope of this article.88 Treatment aims to improve ventricular function and contractility, optimize preload and decrease afterload,85 in order to maximize cardiac output (CO) and tissue perfusion while reducing myocardial oxygen consumption.86
Inotropic agentsContinuous infusion of the beta-adrenergic agonist dobutamine (or low doses of dopamine) is often used to increase contractility and reduce afterload. High dopamine doses should be avoided due to its alpha-adrenergic effect, which may lead to increased afterload. All of these drugs increase myocardial oxygen consumption and are pro-arrhythmic. Prolonged use may adversely affect prognosis.85
DiureticsDiuretics can be used in acute decompensation, in order to increase diuresis and reduce end-diastolic pressure in both ventricles. However, it is important to avoid excessive diuresis, which can compromise preload and thus CO,85 and diuretic therapy should not be excessive or indiscriminate because of potential adverse effects (sodium and potassium depletion, ototoxicity and renal failure).88 Diuretics also play a role in long-term treatment of HF, as discussed below.
Afterload-reducing agentsContinuous infusion of milrinone or amrinone (phosphodiesterase III inhibitors) may be used if further reduction in afterload is required, although systemic hypotension may occur.85,87 These drugs exhibit a mild inotropic effect and mild vasodilatory properties, and increase myocardial relaxation velocity. However, they are also associated with an increased risk of arrhythmias.88 Nitroprusside, a potent vasodilator, can be used for the same purpose, but it has the same potential for causing hypotension, which should be monitored.85 Levosimendan, a calcium-sensitizing agent, increases contractile force and promotes vasodilation by opening vascular potassium channels. Its use has not been proved to reduce mortality, but some reports indicate benefits in weaning from catecholamine support in children with HF.88
Preload- and afterload-reducing agentsNesiritide is a recombinant form of BNP that promotes both diuresis and vasodilation,86,87 and its use is gaining support due to its safety and efficacy in the acute setting.89 Nesiritide directly inhibits the sympathetic nervous system, mineralocorticoid expression and cardiac fibroblast activation, and promotes myocyte survival.86 However, studies in pediatric populations, especially in newborns, are lacking.87
VasopressorsPatients with end-stage HF or vasodilatory shock present with hypotension and can benefit from the cautious use of vasopressors with alpha-adrenergic effects (epinephrine or dopamine). Arginine-vasopressin is also reported to have beneficial hemodynamic effects.85,86,88
Long-term managementTreatment aims to reduce afterload, inhibit the renin-angiotensin system, promote positive ventricular remodeling and optimize preload with diuretic therapy.85 The various approaches that may be involved remain controversial and are based on extrapolation from clinical trials on adults.89
DiureticsThere are no studies that show a reduction in mortality or morbidity with the use of diuretics, but these can be used in patients with fluid retention.87 Use of diuretics alone is not recommended, since they may lead to neurohormonal activation and contribute to disease progression. The most commonly used are loop diuretics (e.g. furosemide), thiazide (e.g. hydrochlorothiazide), and an aldosterone antagonist (spironolactone).6 The aim is to achieve a euvolemic state. However, excessive diuresis may cause intolerance to angiotensin receptor blockers (ARBs) and beta-blockers.89 Patients who remain on diuretic therapy should simultaneously take an angiotensin-converting enzyme (ACE) inhibitor in order to counteract the renin elevation resulting from diuretic therapy.89 Spironolactone, an aldosterone antagonist, appears to reduce the profibrotic effects of aldosterone on the heart and is logical to use in patients taking ACE inhibitors and conventional diuretic agents due to their elevated aldosterone levels.89
Angiotensin-converting enzyme inhibitors and angiotensin receptor blockersThe biological plausibility of ACE inhibitors stems from the fact that activation of the renin-angiotensin system is central to the pathophysiology of HF, regardless of its cause.6 Their main benefit is a reduction in angiotensin II levels, resulting in a drop in systemic vascular resistance.89 A large body of evidence supports their use in almost all forms of HF, including DCM.89 In adults this therapy improves symptoms and reduces hospitalizations and cardiovascular mortality, but these effects are not proven in newborns.6 ARBs have similar biological effects but fewer side effects compared to ACE inhibitors.6 Drugs that block the renin-angiotensin system are indicated in patients with DCM and moderate to severe LV dysfunction regardless of the presence of symptoms, but are not recommended as initial therapy in the presence of decompensated LV dysfunction.6,87 They should be used with caution in neonates because they can easily precipitate renal failure.89
Beta-blockersThe rationale for the use of these agents lies in the fact that excessive sympathetic activity contributes to HF. They are usually well tolerated but may lead to bradycardia, hypotension and fluid retention, and should be avoided in infants with decompensated HF.6 Although widely practiced, their use remains controversial.89 There is evidence that suggests potential improvement in ventricular function and symptoms in children.90 Although their use in adults is supported by good evidence, the only data on infants and children come from small observational studies; there are no specific recommendations for pediatric patients, but their routine use is increasing. The use of carvedilol and metoprolol in pediatric patients is reported in several articles and there has been a multicenter randomized trial in pediatric CM with carvedilol, but its results, although showing a trend for benefit, lack statistical significance.6,90
DigoxinAlthough there is little evidence to support the use of digoxin in pediatric populations, this cardiac glycoside is still widely used in the treatment of HF, for its mild inotropic effect. Its use is restricted to symptomatic HF patients, including those with DCM.6 Some institutional guidelines limit its use to situations in which ventricular rate control is required.89
General management and other treatmentAnticoagulationAnticoagulation is indicated in patients with severe ventricular dysfunction to prevent thromboembolic events.85
Treatment of arrhythmiasArrhythmias are common in patients with DCM.85 However, the negative chronotropic and arrhythmogenic effects of many antiarrhythmic agents limit their use. Amiodarone is reported as safe in the prevention and treatment of arrhythmias, but appears to have no effect on the prevention of sudden death.6,85
Biological immunomodulatorsAlthough it seems to benefit some patient populations in the treatment of adult DCM, to date there is no evidence of benefit of human immunoglobulin in the treatment of neonatal presentation of this disease.91 In cases of acute myocarditis presenting as DCM the use of immunosuppressive regimens which include corticosteroids, azathioprine and cyclosporine has been reported to improve immune-mediated myocardial injury. However, the effectiveness of these therapies has been most evident in chronic virus-negative CM and they therefore appear unpromising in the neonatal period.92
Stem cellsDCM is a myocyte deficiency disorder, since it results from a lack of functioning myocytes (regardless of its etiology). Thus, the possibility of myocardial regeneration by stem cell treatment seems promising, although this therapy is still experimental.93
NutritionEvidence favors increasing the caloric density of feeds as soon as a diagnosis of a heart condition associated with failure to thrive is made. Even in children awaiting surgery, the time before surgical intervention should be used to optimize nutritional status.90 Optimal caloric intake is estimated to be 110-125% of the estimated energy requirement for age and gender.94 Unlike for adults, sodium restriction is not recommended in children.90
Non-pharmacological treatmentHeart transplantation is the mainstay of treatment of end-stage HF.6 Indications for heart transplantation include failure of medical therapy, severe failure to thrive, intractable arrhythmias and severe limitations to activity.85 Routine implantation of a cardiac defibrillator in children with LV dysfunction is not indicated, except for certain types of congenital heart disease.90 Extracorporeal membrane oxygenation and mechanical ventricular assistance devices in children with end-stage heart failure have shown good results as a bridge to recovery or to transplantation.6,86,87 Their use is increasing as devices that allow for smaller pump volumes become available.90
PrognosisApproximately one third of patients with DCM die from the disease, one third continue to have chronic HF, and one third experience improvement in their condition. Causes of death include HF, ventricular arrhythmias and transplantation-related complications. In a small single-center study, DCM was associated with poorer prognosis compared with HCM, as was diagnosis in the neonatal period.95
Children with underlying muscle disorders with progressive dilatation of the heart and worsening HF have a worse prognosis compared with patients with idiopathic DCM.96 If a treatable cause is discovered, prognosis is better. Prognosis is worst for CM secondary to storage diseases that do not have effective therapy. In DCM with no obvious detectable etiology, outcome depends on severity of myocardial dysfunction, improvement during the first year after onset, compliance with therapy, and availability of timely transplant. The degree of depression of fractional shortening or LVEF on initial echocardiography, cardiothoracic ratio and elevation of LV end-diastolic pressure, have all been applied as predictors of outcome, although they are often not predictive. Other possible prognostic factors include age at onset (better for infants), presence of symptomatic arrhythmias, and thromboembolic episodes. A recent review of outcomes from the Pediatric Cardiomyopathy Registry places the incidence of sudden cardiac deaths at 3% and suggests age at diagnosis younger than 14.3 years, LV dilation, and LV posterior wall thinning as predictors of risk.96
Arrhythmic death can occur even after LVEF has returned to normal. Following cardiac transplant, survival rates of as much as 77% at one year and 65% at five years have been reported in children. Mortality and morbidity have greatly decreased because of advances in medical management.
Conflicts of interestThe authors have no conflicts of interest to declare.
