




Dexmedetomidine is a highly selective alpha-2 adrenoceptor agonist that has sedative and analgesic properties and myocardial protective effects. However, the mechanism underlying the protective effect of dexmedetomidine on cardiomyocytes remains unknown. This study mainly aimed to investigate the effects of dexmedetomidine on the generation of reactive oxygen species (ROS) in cardiomyocytes and whether it inhibits the apoptosis of cardiomyocytes by affecting antioxidant enzyme expression.
MethodsNeonatal rat cardiomyocytes were pretreated with dexmedetomidine (100 nM) for 24 h. The cardiomyocytes were then incubated with 200 μM hydrogen peroxide solution (H2O2) for 4 h. PCR assay was used to determine the mRNA expression of antioxidant enzymes. Western blot assay was used to determine the protein expression of antioxidant enzymes. Fluorescence microscopy with the MitoSOX probe was used to detect the formation of ROS in cardiomyocytes, and fluorescence-activated cell sorting with annexin V/PI was used to determine the number of apoptotic cardiomyocytes.
ResultsDexmedetomidine reduced ROS generation and antioxidant enzymes levels in cardiomyocytes before H2O2 stimulation (p<0.05). However, ROS generation and apoptosis in cardiomyocytes were significantly increased after H2O2 treatment, and dexmedetomidine pretreatment markedly inhibited the changes (p<0.05).
ConclusionFor the first time, to the best of our knowledge, our study shows that dexmedetomidine has a protective effect on cardiomyocytes through inhibition of ROS-induced apoptosis, and more importantly, this effect is independent of antioxidant enzyme mRNA and protein expression.
A dexmedetomidina é um agonista altamente seletivo do adrenoreceptor α2, que tem propriedades sedativas e analgésicas e efeitos protetores miocárdicos. No entanto, o mecanismo subjacente ao efeito protetor da dexmedetomidina nos cardiomiócitos permanece desconhecido. Este estudo teve como objetivo principal investigar os efeitos da dexmedetomidina na geração de espécies reativas de oxigénio (ROS) em cardiomiócitos e se a inibição da apoptose de cardiomiócitos pode afetar a expressão de enzimas antioxidantes.
MétodosCardiomiócitos de ratos neonatais foram pré-tratados com dexmedetomidina (100nM) por 24h. Em seguida, os cardiomiócitos foram incubados com solução de peróxido de hidrogênio (H2O2) 200μM por 4h. O teste de PCR foi usado para determinar a expressão de mRNA de enzimas antioxidantes. O teste de Western blot foi usado para determinar a expressão de proteínas de enzimas antioxidantes. A microscopia de fluorescência com a sonda MitoSOX foi aplicada para detetar a formação de ROS em cardiomiócitos e classificação de células ativadas por fluorescência (FACS) com Anexina V/PI foi usada para detetar o número de cardiomiócitos apoptóticos.
ResultadosA dexmedetomidina reduziu a geração de ROS e os níveis de enzimas antioxidantes em cardiomiócitos antes da estimulação com H2O2 (P<0,05). No entanto, a geração de ROS e apoptose em cardiomiócitos aumentou significativamente após o tratamento com H2O2, o pré-tratamento com dexmedetomidina inibiu notavelmente as alterações (P<0,05).
ConclusãoPela primeira vez, até onde sabemos, o nosso estudo identifica que a dexmedetomidina tem um efeito protetor sobre os cardiomiócitos por meio da inibição da apoptose induzida por ROS e, mais importante, esse efeito é independente do RNAm da enzima antioxidante e da expressão de proteínas.
Dexmedetomidine, a highly selective alpha 2-adrenergic agonist, is notable for its ability to alleviate the stress response associated with the perioperative period and to prevent postoperative cardiac complications.1 It can reduce postoperative mortality and improve prognosis of patients undergoing cardiac surgery.2 Dexmedetomidine has been shown to inhibit cardiac ischemia-reperfusion injury via the endothelial nitric oxide synthase/nitric oxide (NO) signaling pathway in rats.3,4 Previous research has also shown that dexmedetomidine inhibits reactive oxygen species (ROS)-induced cardiomyocyte apoptosis by reducing mitochondrial response sensitivity.5 However, it is unclear whether antioxidant enzymes are involved in the cardioprotective effect of dexmedetomidine.
ROS are generated during electron transfer in the mitochondrial respiratory chain, and complex I and complex III are the main sources of ROS generated in the body.6,7 Under normal physiological conditions, ROS are important in cellular homeostasis and the immune response. However, excessive ROS caused by high metabolism have adverse effects, such as DNA degeneration and gene transcription activation, leading to tumor formation and inflammation.8,9 In addition, ROS are linked to metabolic heart disease, by causing oxidative modification of complex I and complex I subunit proteins, leading to mitochondrial dysfunction.10 Dhalla et al. report that ROS can also induce apoptosis and are implicated in the pathophysiology of various cardiovascular diseases.11 The relative equilibrium state of ROS in vivo is determined by the production rate of ROS and the clearance rate of antioxidants. Antioxidant enzymes, including superoxide dismutase 2 (SOD2), glutathione peroxidase 4 (GPX4), glutaredoxin (Grx1) and catalase, are crucial antioxidants that clear the toxic effects of excessive ROS in vivo.12–14 Thus, reduced levels of antioxidant enzymes can lead to redox imbalance and irreversible cell damage.15,16
Previous studies have confirmed that dexmedetomidine inhibits the sensitivity of mitochondria to H2O2 stimulation in cardiomyocytes by preventing the generation of ROS, thereby suppressing ROS-induced cardiomyocyte apoptosis. The decrease in ROS levels is mainly due to dexmedetomidine inhibiting genes involved in mitochondrial biosynthesis and mitochondrial respiratory chain complex-related protein expression.5 Taking into account the current literature on the subject, this study aims to further investigate the effect of dexmedetomidine on ROS-induced cardiomyocyte apoptosis, and to determine whether these effects are reflected in antioxidant protein transcription and expression.
MethodsReagents and groupsDexmedetomidine was purchased from Sigma-Aldrich, Merck KGaA (Darmstadt, Germany) and was added to Dulbecco's modified Eagle medium (DMEM) to obtain the desired concentration. H2O2 solution and collagenase type II were obtained from Sigma-Aldrich, Merck KGaA (Darmstadt, Germany). The primary rat cardiomyocytes were cultured with dexmedetomidine stimulation for 24 h, and were then divided into a control group and a 100 nM dexmedetomidine group. After 200 μM H2O2 stimulation for 4 h, fluorescence microscopy with the MitoSOX probe was used to detect the generation of ROS with and without H2O2 stimulation, and cardiomyocyte apoptosis was detected by fluorescence-activated cell sorting (FACS). The expression of antioxidant enzymes was detected by polymerase chain reaction (PCR).
Cell cultureNeonatal Wistar rat cardiomyocytes were cultured as reported previously.5,17 Whole hearts of neonatal Wistar rats (1-3 days) were purchased from Shanghai Laboratory Animal Center (SLAC, Shanghai, China), cut into 1-2 mm sections and digested in a digestion solution (0.025% type II collagen, 0.06% trypsin, and 20 μg/ml DNase) at 37°C three times, for 15 min each time. The homogenate was loaded onto a 45.5% Percoll gradient over another 58.5% Percoll gradient, followed by centrifugation at 15°C and 3800 g for 30 min. The dissociated cardiomyocytes were collected and seeded at 1×105/cm2, followed by culture in DMEM (Thermo Fisher Scientific, Inc., Waltham, MA, USA) with 10% fetal bovine serum (Thermo Fisher Scientific, Inc.) at 37°C with 5% CO2 and 95% O2. In all experiments, 100 nM of dexmedetomidine (Sigma, Merck Millipore, Darmstadt, Germany) was used for cardiomyocyte treatment at 37°C for 24 h. The dose of dexmedetomidine used in the present study was based on a previous study5 and our preliminary experiments. All experiments were approved by the Animal Care and Use Committee of Shanghai General Hospital (Shanghai, China) and performed in accordance with the relevant guidelines and regulations of the Bio-X Institutes of Shanghai Jiao Tong University (Shanghai, China).
MitoSOX assaysA MitoSOX probe was applied to detect the generation of ROS in neonatal Wistar rat cardiomyocytes. The primary neonatal rat cardiomyocytes were cultured in collagen-coated 96-well dishes. A live-cell permeable probe, MitoSOX (Invitrogen; Thermo Fisher Scientific, Inc.), was used following dexmedetomidine stimulation for 24 h. For the MitoSOX assay, on the day of measurement the cells were washed twice in sterilized phosphate-buffered saline (PBS) followed by stimulation with 200 μM of H2O2 at 37°C for 4 h. The cells were then incubated in the dark with 5 μM MitoSOX-containing Hanks’ Balanced Salt Solution (HBSS) medium at 37°C for 10 min and were measured at ex/em: 510/580 nm after being washed three times with PBS. The cells were observed by confocal microscopy after smear.
Fluorescence-activated cell sorting analysisFACS analysis was performed to detect H2O2-induced cardiomyocyte apoptosis, as reported previously.18 Briefly, the primary rat cardiomyocytes were cultured in 6-well dishes. For cellular apoptosis, H2O2 (200 μM) was added to the cells and incubated at 37°C for 4 h, following exposure of the cells to dexmedetomidine pretreatment for 24 h. The cardiomyocytes (2×106 cells/ml) were labeled fluorescently for the detection of apoptotic and necrotic cells by adding 50 μl 1×binding buffer and 5 μl annexin V-FITC (BD Pharmingen, San Diego, CA, USA) and 2 μl propidium iodide (Cedarlane Laboratories, Hornby, ON, Canada). The samples were incubated in the dark at room temperature for 15 min following gentle mixing. A minimum of 20000 cells within a gated region were analyzed using FACS analysis (Epics Altra flow cytometer; Beckman Coulter, Fullerton, CA, USA).
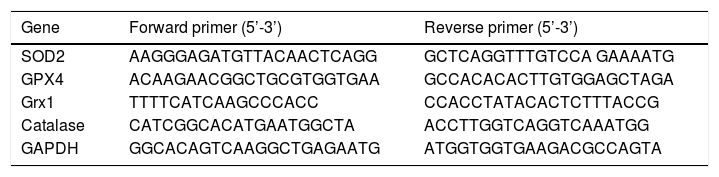
Quantitative real-time polymerase chain reaction analysisTotal RNA was extracted from the cultured cardiomyocytes using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. Complementary DNA (cDNA) (50 ng/μl) was synthesized using oligo (dT) primers with the Transcriptor First Strand cDNA Synthesis kit (Roche Diagnostics, Shanghai, China). The real-time PCR amplifications were quantified using SYBR-Green (Roche Diagnostics) and the reaction system was composed of 10 μl SYBR premix Ex Taq II, 0.2 μl primers, 8.8 μl H2O, and 1 μl cDNA. The final concentration of cDNA used in this experiment was 2.5 ng/μl. The thermocycling conditions were as follows: 5 s at 95°C followed by 30 s at 60°C for 42 cycles using a real-time thermal cycler dice system (Takara Biotechnology Co., Ltd.). The levels of relative mRNA expression were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and data analysis was performed using the 2-ΔΔCt method. All experiments were repeated at least in triplicate. The primers used in the present study are listed in Table 1.
Primers used in real-time fluorescence quantitative polymerase chain reaction.
| Gene | Forward primer (5’-3’) | Reverse primer (5’-3’) |
|---|---|---|
| SOD2 | AAGGGAGATGTTACAACTCAGG | GCTCAGGTTTGTCCA GAAAATG |
| GPX4 | ACAAGAACGGCTGCGTGGTGAA | GCCACACACTTGTGGAGCTAGA |
| Grx1 | TTTTCATCAAGCCCACC | CCACCTATACACTCTTTACCG |
| Catalase | CATCGGCACATGAATGGCTA | ACCTTGGTCAGGTCAAATGG |
| GAPDH | GGCACAGTCAAGGCTGAGAATG | ATGGTGGTGAAGACGCCAGTA |
GAPDH: glyceraldehyde 3-phosphate dehydrogenase; GPX4: glutathione peroxidase 4; Grx1: glutaredoxin 1; SOD2: superoxide dismutase 2.
The cultured cardiomyocytes were scraped and collected, and the cardiomyocytes, centrifuged at 1500 g at 4°C for 5 min, were dissolved in radioimmunoprecipitation assay buffer (RIPA) containing 50 mM Tris, pH 7.4, 150 mM NaCl, 1% NP40, 0.5% Na+ deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and protease inhibitor cocktails (Sigma Aldrich, St. Louis, MO, USA). The samples containing equal protein as determined by a micro-bicinchoninic acid (BCA) assay kit (Beyotime) were diluted in loading buffer. Total protein (10 μg) was loaded and separated using SDS-PAGE (8-20% gel) for 90 min, and then transferred onto polyvinylidene fluoride membranes for another 90 min. Following blocking in 5% skim milk for 1 h, the membranes were incubated in primary antibodies at 4°C overnight. The unbound antibodies were then washed off in TBST 3-5 times, followed by incubation with horseradish peroxidase-conjugated sheep anti-rabbit immunoglobulin G (1:2000; Bio-Techne Ltd., Oxford, UK) at room temperature for 1 h. The FluorChem E imaging system (Cell Biosciences, Inc., Shanghai, China) was used to visualize the signals. The primary antibodies used were as follows: SOD2 (1:1000; Cell Signaling Technology, MA, USA); GPX4 (1:1000; Cell Signaling Technology, MA, USA); Grx1 (1:2000; Cell Signaling Technology, MA, USA); catalase (1:2000; Cell Signaling Technology, MA, USA) and beta-actin (1:2000; Cell Signaling Technology, MA, USA). Densitometric analysis of each band was conducted with ImageJ software. Beta-actin was used as an internal loading control.
Statistical analysisAll experiments were repeated two or three times. All results are reported as mean±standard error of the mean. Comparisons between two groups were analyzed using the Student's t test. Multiple comparisons between groups were performed using one-way analysis of variance with Tukey's multiple comparisons test (IBM SPSS 20.0 and GraphPad Prism 6.0). p<0.05 was considered to indicate a statistically significant difference.
ResultsDexmedetomidine down-regulates reactive oxygen species levels in cardiomyocytesIn the present study, ROS levels in neonatal rat cardiomyocytes were measured both under normal conditions and following H2O2 stimulation. Dexmedetomidine was shown to significantly reduce cardiomyocyte ROS levels under normal conditions compared with cardiomyocytes untreated with dexmedetomidine (p<0.01). After H2O2 was added to cardiomyocytes untreated with dexmedetomidine, ROS levels were markedly increased. However, the upregulated ROS levels were dramatically inhibited by dexmedetomidine preconditioning (p<0.001) (Figure 1).
 Relative MitoSOX fluorescence of cardiomyocytes treated by dexmedetomidine with or without H2O2 (n=8 per group); (B) representative MitoSOX fluorescence images of cardiomyocytes with or without H2O2 stimulation following dexmedetomidine pretreatment (scale bar=50 μm). Statistical significance was determined using one-way analysis of variance (**p<0.01, ***p<0.001, 100 nM vs. control, n=10 per group). 100 nM group: cardiomyocytes treated with dexmedetomidine; Dex: dexmedetomidine; H2O2: hydrogen peroxide; ROS: reactive oxygen species.")
Dexmedetomidine decreases sensitivity of ROS response to H2O2 in cardiomyocytes. (A) Relative MitoSOX fluorescence of cardiomyocytes treated by dexmedetomidine with or without H2O2 (n=8 per group); (B) representative MitoSOX fluorescence images of cardiomyocytes with or without H2O2 stimulation following dexmedetomidine pretreatment (scale bar=50 μm). Statistical significance was determined using one-way analysis of variance (**p<0.01, ***p<0.001, 100 nM vs. control, n=10 per group). 100 nM group: cardiomyocytes treated with dexmedetomidine; Dex: dexmedetomidine; H2O2: hydrogen peroxide; ROS: reactive oxygen species.
The effect of dexmedetomidine on H2O2-induced cardiomyocyte apoptosis was investigated through FACS assay, the results of which are shown in Figure 2. First, the cardiomyocytes were exposed to 100 nM of dexmedetomidine and it was demonstrated that this pretreatment significantly attenuated the loss of cardiomyocytes induced by H2O2. However, the apoptosis rate of cardiomyocytes without dexmedetomidine pretreatment was significantly increased by H2O2 stimulation. These results suggest that dexmedetomidine pretreatment created protective cellular conditions, which resisted ROS-induced apoptosis and increased cell survival.
 analysis. (A) Cardiomyocyte apoptosis was determined by analyzing annexin V and propidium iodide binding with FACS; (B) quantification of the apoptotic cardiomyocyte percentage. Statistical significance was determined using one-way analysis of variance (*p<0.05, H2O2 vs. control; **p<0.01, Dex+H2O2 vs. H2O2; ***p<0.001, Dex+H2O2 vs. control, n=10 per group). H2O2 group: cardiomyocytes induced by H2O2 without dexmedetomidine preconditioning; Dex+H2O2 group: cardiomyocytes induced by H2O2 with dexmedetomidine preconditioning. Dex: dexmedetomidine; H2O2: hydrogen peroxide.")
Dexmedetomidine suppresses H2O2-induced cardiomyocyte apoptosis. Apoptosis was detected using fluorescence-activated cell sorting (FACS) analysis. (A) Cardiomyocyte apoptosis was determined by analyzing annexin V and propidium iodide binding with FACS; (B) quantification of the apoptotic cardiomyocyte percentage. Statistical significance was determined using one-way analysis of variance (*p<0.05, H2O2 vs. control; **p<0.01, Dex+H2O2 vs. H2O2; ***p<0.001, Dex+H2O2 vs. control, n=10 per group). H2O2 group: cardiomyocytes induced by H2O2 without dexmedetomidine preconditioning; Dex+H2O2 group: cardiomyocytes induced by H2O2 with dexmedetomidine preconditioning. Dex: dexmedetomidine; H2O2: hydrogen peroxide.
To further investigate the effect of dexmedetomidine on antioxidant enzyme expression in cardiomyocytes, mRNA and protein expression of SOD2, GPX4, Grx1 and catalase were determined in neonatal rat cardiomyocytes. Dexmedetomidine treatment significantly down-regulated mRNA and protein levels of Grx1 and mRNA level of GPX4. While mRNA and protein levels of SOD2 were marginally reduced and protein level of catalase was increased, these differences were not statistically significant (Figures 3 and 4).
, glutathione peroxidase 4 (GPX4), glutaredoxin 1 (Grx1), and catalase in cardiomyocytes. mRNA expression of antioxidant enzymes was detected by real-time fluorescence quantitative polymerase chain reaction (RT-PCR) analysis. Relative gene expression levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Statistical significance was determined using one-way analysis of variance (*p<0.05, **p<0.01, 100 nM vs. control, n=10 per group). 100 nM group: cardiomyocytes treated with dexmedetomidine; H2O2: hydrogen peroxide.")
Dexmedetomidine suppresses mRNA expression of superoxide dismutase 2 (SOD2), glutathione peroxidase 4 (GPX4), glutaredoxin 1 (Grx1), and catalase in cardiomyocytes. mRNA expression of antioxidant enzymes was detected by real-time fluorescence quantitative polymerase chain reaction (RT-PCR) analysis. Relative gene expression levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Statistical significance was determined using one-way analysis of variance (*p<0.05, **p<0.01, 100 nM vs. control, n=10 per group). 100 nM group: cardiomyocytes treated with dexmedetomidine; H2O2: hydrogen peroxide.
, glutathione peroxidase 4 (GPX4), glutaredoxin 1 (Grx1), and catalase in cardiomyocytes. Protein expression of antioxidant enzymes was detected by Western blot analysis. Relative protein expression levels were normalized to beta-actin. Statistical significance was determined using one-way analysis of variance (*p<0.05, **p<0.01, 100 nM vs. control, n=10 per group). 100 nM group: cardiomyocytes treated with dexmedetomidine; H2O2: hydrogen peroxide.")
Dexmedetomidine suppresses protein expression of superoxide dismutase 2 (SOD2), glutathione peroxidase 4 (GPX4), glutaredoxin 1 (Grx1), and catalase in cardiomyocytes. Protein expression of antioxidant enzymes was detected by Western blot analysis. Relative protein expression levels were normalized to beta-actin. Statistical significance was determined using one-way analysis of variance (*p<0.05, **p<0.01, 100 nM vs. control, n=10 per group). 100 nM group: cardiomyocytes treated with dexmedetomidine; H2O2: hydrogen peroxide.
In the present study, we investigated the effect of dexmedetomidine on ROS generation and apoptosis in cardiomyocytes and determined the associated changes in the expression of SOD2, GPX4, Grx1 and catalase. For the first time, our results demonstrated that dexmedetomidine preconditioning of cardiomyocytes decreased ROS generation in cardiomyocytes, thus protecting against ROS-induced apoptosis. More importantly, we found the protective effect of dexmedetomidine on cardiomyocytes was not reflected in protein transcription and expression of antioxidant enzymes.
Multiple mechanisms have been suggested to contribute to the organ-protective effects of dexmedetomidine.19–21 For example, dexmedetomidine-induced cardioprotection may be related to activation of the PI3K-AKT pro-survival pathway or inhibition of the HMGB1-TLR4-NFκB inflammatory pathway.21,22 It is well known that oxidative stress is closely linked to cardiac dysfunction.23,24 Liu et al. reported that dexmedetomidine attenuates H2O2-induced ROS generation and consequently inhibits neonatal rat cardiomyocyte apoptosis.25 In addition, dexmedetomidine alleviates lipopolysaccharide-induced acute lung injury by inhibiting oxidative stress.20 These previous studies indicate that oxidative stress signaling is a key target for the beneficial effects of dexmedetomidine. It is well known that oxidative stress occurs when ROS generation exceeds antioxidant capacity.26 In the present study, along with myocardial injury, ROS generation was, predictably, increased, which was dramatically inhibited by dexmedetomidine. In addition, a significant decrease was observed in the expression of antioxidant enzymes in cardiomyocytes pretreated with dexmedetomidine. These findings demonstrated that the protective effect of dexmedetomidine on cardiomyocytes was independent of antioxidant enzyme expression levels.
According to the new concept of ROS-induced ROS release, exposure to excessive oxidative stress results in increased ROS reaching a threshold level that triggers the opening of the mitochondrial permeability transition pore or inner membrane anion channel. In turn, this leads to the simultaneous collapse of the mitochondrial membrane potential and transient increased ROS generation by the electron transfer chain. These generated ROS can be released into the cytosol, inducing neighboring mitochondria to generate more ROS.27,28 In this case, ROS have a signal transduction function, expanding local mitochondrial damage and causing cell damage through this positive feedback mechanism. It should be emphasized that the premise of this positive feedback is that mitochondria are highly sensitive to ROS stimulation, which will cause a series of changes in membrane channels, mitochondrial membrane potentials and the respiratory chain complex.27,28 In the present study, dexmedetomidine attenuated H2O2-induced increase in intracellular ROS generation, and therefore reduced the number of apoptotic cardiomyocytes. However, the expression of antioxidant enzymes was decreased in cardiomyocytes pretreated with dexmedetomidine, indicating that decreased mitochondrial sensitivity is a key factor behind dexmedetomidine's inhibition of the increase of ROS levels in cardiomyocytes.5
The mechanism of dexmedetomidine's protective effect against increased ROS levels in cardiomyocytes is not clear. Interestingly, according to our observations, dexmedetomidine markedly decreased the expression of antioxidant enzymes in cardiomyocytes, unlike in previous studies. Li et al.29 found that dexmedetomidine significantly increased SOD2 and catalase proteins and the ability to scavenge oxygen free radicals, and improved ventilation and increased blood oxygen partial pressure in rats with chronic obstructive pulmonary disease. Wang et al.30 investigated whether cotreatment with dexmedetomidine and Na2SeO3 improved antioxidant and anti-inflammatory protection of myocardial cells from ischemia/reperfusion injury compared to their individual treatments. They found that treatment with dexmedetomidine increased SOD1, SOD2, GPx1, and GPx2 mRNA expression in H9C2 cells in oxygen-glucose deprivation/reoxygenation. This inconsistency may be due to differences in histological techniques, methods for detecting antioxidant enzymes, or dosages and durations of dexmedetomidine intervention; there may be other possible explanations.
In vascular endothelial cells, PGC-1alpha participates in mitochondrial biosynthesis and oxidative phosphorylation, regulates mitochondrial antioxidant enzyme transcription, and increases expression of SOD, catalase, Prx3, Prx5, TRX2, and TRXR2, which reduces ROS accumulation and the mitochondrial stress response.14 In addition, PGC-1alpha also inhibits the neurological damage caused by ROS by activating the expression of antioxidant enzymes such as SOD, GPX and catalase.13 The expression of PGC-1alpha, PPAR, NRF1 and ERRɑ is significantly reduced in cardiomyocytes treated with dexmedetomidine.5 The decrease in antioxidant enzymes in cardiomyocytes may be related to the decreased expression of PGC-1alpha. It has been reported that carvedilol inhibits ROS-induced myocardial apoptosis by activating SOD and GPX mechanisms.31 Grx1 suppresses NO-induced apoptosis in cardiomyocyte by inhibiting oxidative modification.12 However, it is unclear whether the decrease in antioxidant enzymes is accompanied by adverse effects. Studies have found that dexmedetomidine significantly attenuates the increase in ROS and apoptosis in H2O2-stimulated cardiomyocytes without affecting mitochondrial respiratory function.5 This shows that ROS are still in a relatively balanced state, and the decline in the expression of these antioxidant enzymes does not affect ROS equilibrium or promote apoptosis. Moreover, the redox state is a dynamic equilibrium process, and overexpressed antioxidant enzymes can also cause reduction stress, causing toxic effects.32 Therefore, dexmedetomidine attenuates the increase of ROS levels in cardiomyocytes and inhibits ROS-induced apoptosis, which is independent of antioxidant enzyme expression. However, the mechanisms by which dexmedetomidine inhibits antioxidant enzyme levels in cardiomyocytes still need further study.
ConclusionIn summary, the present study shows that preventive treatment with dexmedetomidine protects neonatal rat cardiomyocytes against H2O2-induced ROS generation and apoptosis. More importantly, the protective effect of dexmedetomidine is independent of antioxidant enzyme protein transcription and expression. The scope of this study was limited in terms of the precise mechanism of dexmedetomidine in oxidative stress and apoptosis, which will be investigated in future studies.
Data availabilityThe data used to support the findings of this study are available from the corresponding author on reasonable request.
Funding statementThis study was supported by a grant from the National Natural Science Foundation of China (grant number 81171845).
Conflicts of interestThe authors have no conflicts of interest to declare.
The authors would like to thank all their colleagues who contributed to this study.

www.publicationethics.org.