





Sneddon syndrome is a rare clinical entity characterized by the association of ischemic cerebrovascular disease and livedo reticularis. The authors report a case of stroke and myocardial infarction in a 39-year-old man with Sneddon syndrome and antiphospholipid syndrome who subsequently met some criteria for systemic lupus erythematosus, highlighting the complexity of cardiovascular involvement in systemic diseases.
A síndroma de Sneddon é uma entidade rara caracterizada pela associação de doença cerebrovascular isquémica e livedo reticularis. Os autores apresentam um caso de acidente vascular cerebral isquémico e enfarte do miocárdio em doente com síndroma de Sneddon e síndroma antifosfolipídica, que posteriormente reuniu alguns critérios de lúpus eritematoso sistémico, salientando a complexidade do envolvimento cardiovascular nas doenças sistémicas.
antinuclear antibodies
antiphospholipid antibodies
antiphospholipid syndrome
computed tomography
implantable cardioverter-defibrillator
international normalized ratio
livedo reticularis
left ventricular
left ventricular ejection fraction
myocardial infarction
right middle cerebral artery
magnetic resonance imaging
mitral valve
New York Heart Association
right coronary artery
systemic lupus erythematosus
Sneddon syndrome
transesophageal echocardiography
transthoracic echocardiography
Sneddon syndrome (SS) is a rare clinical entity, with an estimated annual incidence of 4/1 000 000, that mainly affects women aged between 20 and 42 years, although cases have occasionally been reported in childhood and after the age of 65.1 First described by Sneddon in 1965, the syndrome is characterized by the association of ischemic cerebrovascular disease and livedo reticularis (LR), caused by progressive occlusion of small- and medium-caliber arteries.1–3 Organs and systems affected include the cardiovascular system, leading to hypertension, myocardial ischemia and valve damage, most frequently mitral but also aortic, with or without regurgitation.1,2 There may also be involvement of other systems, particularly ocular (50–70%), gastrointestinal and renal (50–70%), as well as venous occlusions. No specific marker has been found for SS and its etiology is unclear,1–8 but it has been suggested that its manifestations result from reduced blood flow due to increased viscosity, thrombotic and embolic events, arterial wall calcification, vasculitis and intimal hyperplasia, resulting in proliferation, recanalization and thrombosis of small- and medium-caliber arteries.6
Although SS was not initially diagnosed in the context of systemic diseases, the presence of antiphospholipid antibodies (APA) has been described in SS patients, with a highly variable prevalence in different series (0–85%). Criteria for systemic lupus erythematosus (SLE) and coagulation disorders including protein S deficiency and factor V Leiden have also been reported.1–5 Three variants of the syndrome are now recognized: an idiopathic form, which is not accompanied by APA or SLE; a primary antiphospholipid syndrome (APS)-related form; and an SLE-associated form, with or without the presence of APA.4
A diagnosis of APS is based on the presence of at least one clinical criterion (documented recurrent arterial and/or venous thromboembolism or recurrent abortions) and one laboratory criterion (lupus anticoagulant or phospholipid-dependent antibodies, anticardiolipin and anti-β2-glycoprotein-I, on at least two occasions at least 12 weeks apart).9,10 Among its cardiovascular manifestations are thrombotic coronary disease, intracardiac thrombi and valve abnormalities (thickening, attached thrombotic masses, and non-infective vegetations). It is more common in women.
SLE is a systemic autoimmune disease, also more common in women, with a prevalence of 40/1 000 000.11 Diagnosis is based on the presence of at least four of the 11 American College of Rheumatology criteria, recently updated to 17 criteria including biopsy-proven lupus nephritis together with antinuclear antibodies (ANA) or anti-double-stranded DNA antibodies.11,12 It is associated with APS in 10–30% of cases, which raises the risk of acute coronary events 10-fold.9 Ischemic coronary disease is one possible cardiac manifestation of SLE, caused by accelerated atherosclerosis, although it may also result from thrombosis in the context of APS due to embolization of non-infective vegetations or, less often, coronary arteritis. Other cardiac manifestations of SLE include pericarditis, pericardial effusion, myocarditis and Libman-Sacks endocarditis.13
Case reportA 39-year-old Caucasian man with a history of smoking (24 pack/years) and hypercholesterolemia and a first-degree relative (mother) with Sjögren syndrome, and no history of corticosteroid therapy, anabolic steroid use or drug abuse, was admitted to the neurology department on January 21, 2010 for ischemic stroke with left hemiparesis. Physical examination revealed extensive livedo reticularis (LR), predominantly on the trunk and upper limbs. No fever or atrial fibrillation was detected. On admission head computed tomography (CT) was performed, which showed a slight subcortical hypodensity in the right lateral frontal region suggesting a recent vascular event, and several small hypodensities in the subcortical white matter in both hemispheres, presumably sequelae of the vascular event. One week later brain magnetic resonance imaging (MRI) to investigate a possible demyelinating etiology showed a recent infarction in the territory of the right middle cerebral artery (RMCA); MRI angiography showed occlusion of a branch of the RMCA, confirming that the lesions were sequelae of the vascular event.
Laboratory tests on admission showed thrombocytopenia (52–54×103 platelets/μl, normal: 150–400×103/μl), erythrocyte sedimentation rate 34 mm/h (normal: <10 mm/h), strongly positive lupus anticoagulant, IgG >280 U/ml and IgM 82 U/ml anticardiolipin antibodies (positive: >30 U/ml), IgG 13 U/ml (positive >12 U/ml) and IgM 46 U/ml (positive >7 U/ml) anti-β2-glycoprotein-I antibodies, anti-mitochondrial antibodies 320 U/ml (positive >20 U/ml), C3 0.786 g/l (normal: 0.9–1.8 g/l) and C4 0.049 g/l (normal: 0.10–0.40 g/l) hypocomplementemia, LDL cholesterol 3.01 mmol/l (normal <2.50 mmol/l), C-reactive protein 0.8 mg/dl (normal: <1 mg/dl) and NT-proBNP 571 pg/ml (elevated >1800 pg/ml). No other abnormalities were observed in blood tests, renal or liver function tests, other antibodies, thrombophilia screen, bacterial or viral serology including HBV, HCV and HIV, tumor markers or toxicology screening. Carotid and vertebral Doppler ultrasound study three days after admission detected no atherosclerotic plaques.
The patient's neurological deficits resolved completely during hospitalization and he was discharged with a diagnosis of SS associated with APS, medicated with warfarin for a target international normalized ratio (INR) of 2–3 and was advised to quit smoking. Transthoracic (TTE) and transesophageal (TEE) echocardiograms were also requested and the patient was referred to the cardiology department to investigate a cardioembolic etiology of the stroke, given his age and the location of the cerebral lesions.
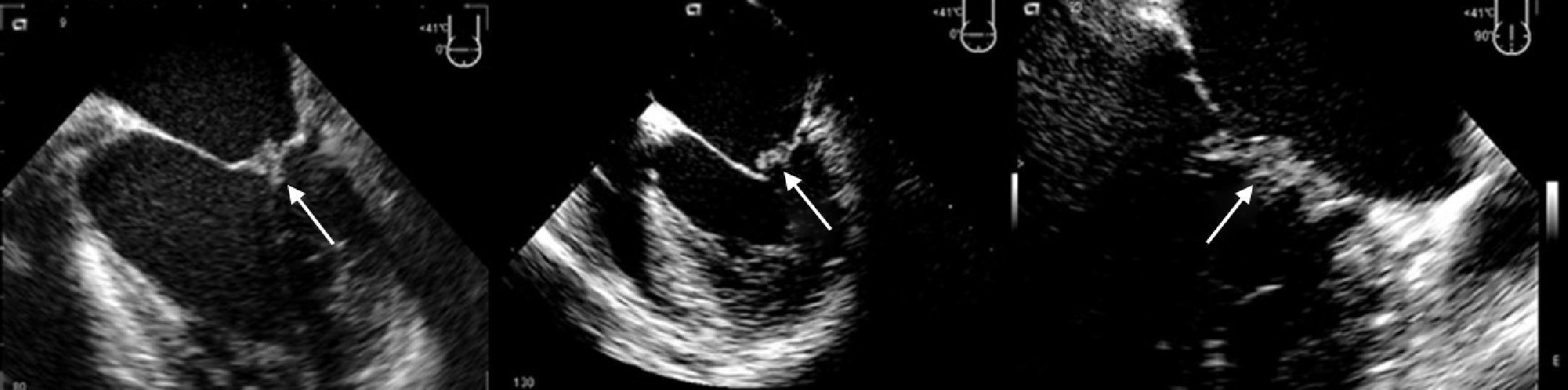
TTE performed a week after admission showed thickening of both mitral valve (MV) leaflets and a small (0.8 cm×0.9 cm) hyperechogenic, homogeneous mass on the atrial face of the posterior leaflet, mild mitral regurgitation, moderate left ventricular (LV) dilatation with wall motion abnormalities (akinesia of the basal and mid segments of the inferior and inferolateral walls, hypokinesia of the apical segments of the inferoseptal wall) and moderate global systolic dysfunction (LV ejection fraction [LVEF] 34% by Simpson's biplane method). TEE on the same day excluded other cardioembolic sources and revealed the MV mass to be sessile, with irregular but well-defined borders, of intermediate echogenicity, heterogeneous and mobile, moving with the leaflets (Figure 1). No spontaneous contrast was observed in the left chambers.
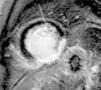
The patient was referred for cardiac MRI to investigate the LV dysfunction, which was performed on May 14. This confirmed severe systolic dysfunction (LVEF 33%); late gadolinium enhancement suggested an ischemic etiology, with transmural fibrosis visible in the inferior and inferolateral walls (Figure 3). T2-weighted imaging also showed a hypointense nodule on the posterior MV leaflet which was too small to be adequately characterized by MRI.
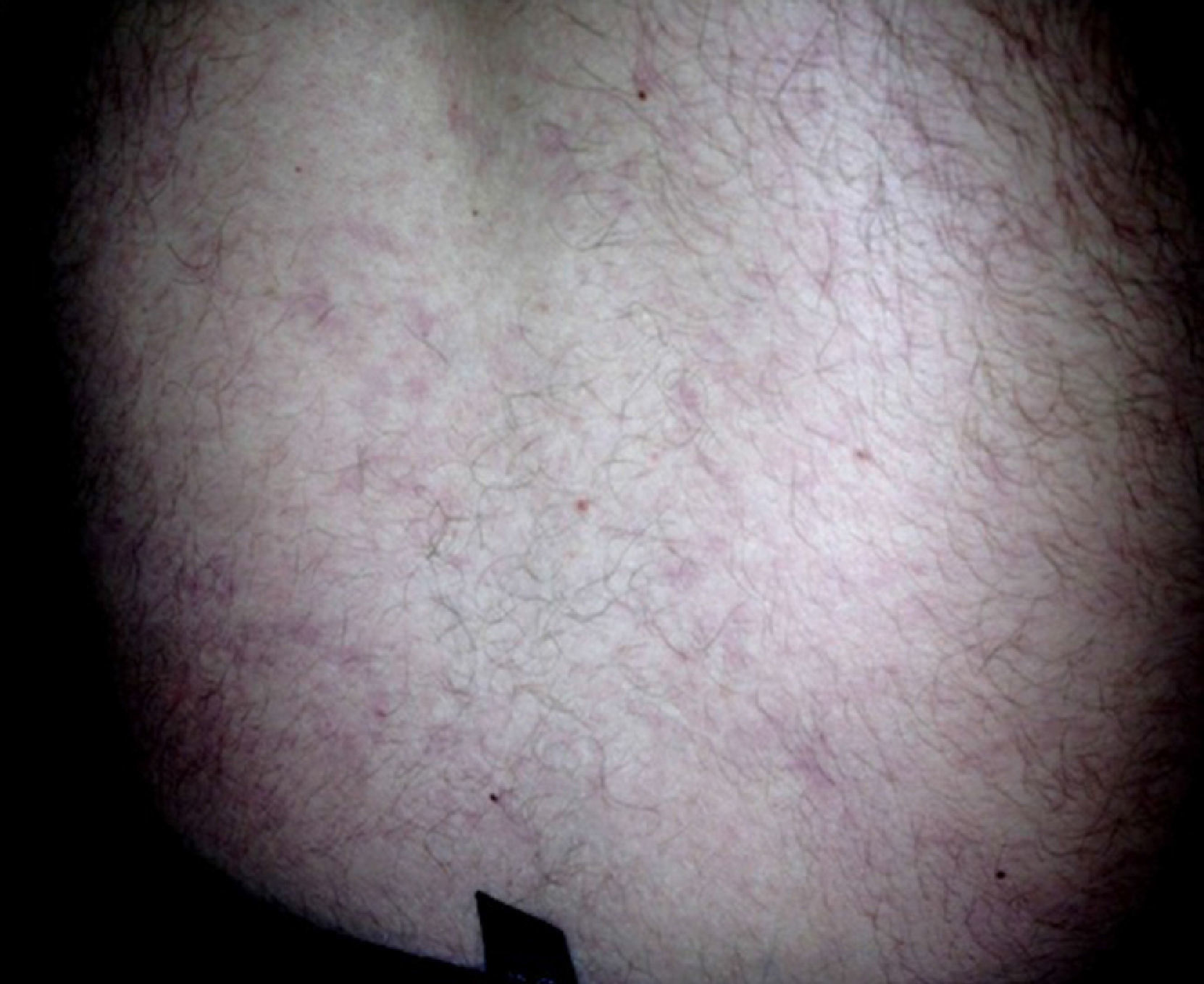
At the first cardiological consultation, four months after discharge (May 24), the patient reported no symptoms apart from a single episode of typical angina three weeks previously, triggered by exertion and lasting for five minutes, which resolved spontaneously after 30 minutes’ rest; he did not seek medical help. He reported arthralgia in the proximal interphalangeal joints of both hands and a history of photosensitivity (although not recent). Physical examination revealed LR, purplish in color, with irregular borders, on the lower half of the trunk (Figure 2), which according to the patient he had had “for a long time” and was now less marked than in the acute phase of the stroke. No other alterations were observed, including neurological sequelae. The electrocardiogram showed sinus rhythm at 70 bpm and Q and negative T waves in the inferior leads. The patient had brought records of INR for the previous two months, with values between 3.2 and 3.4.
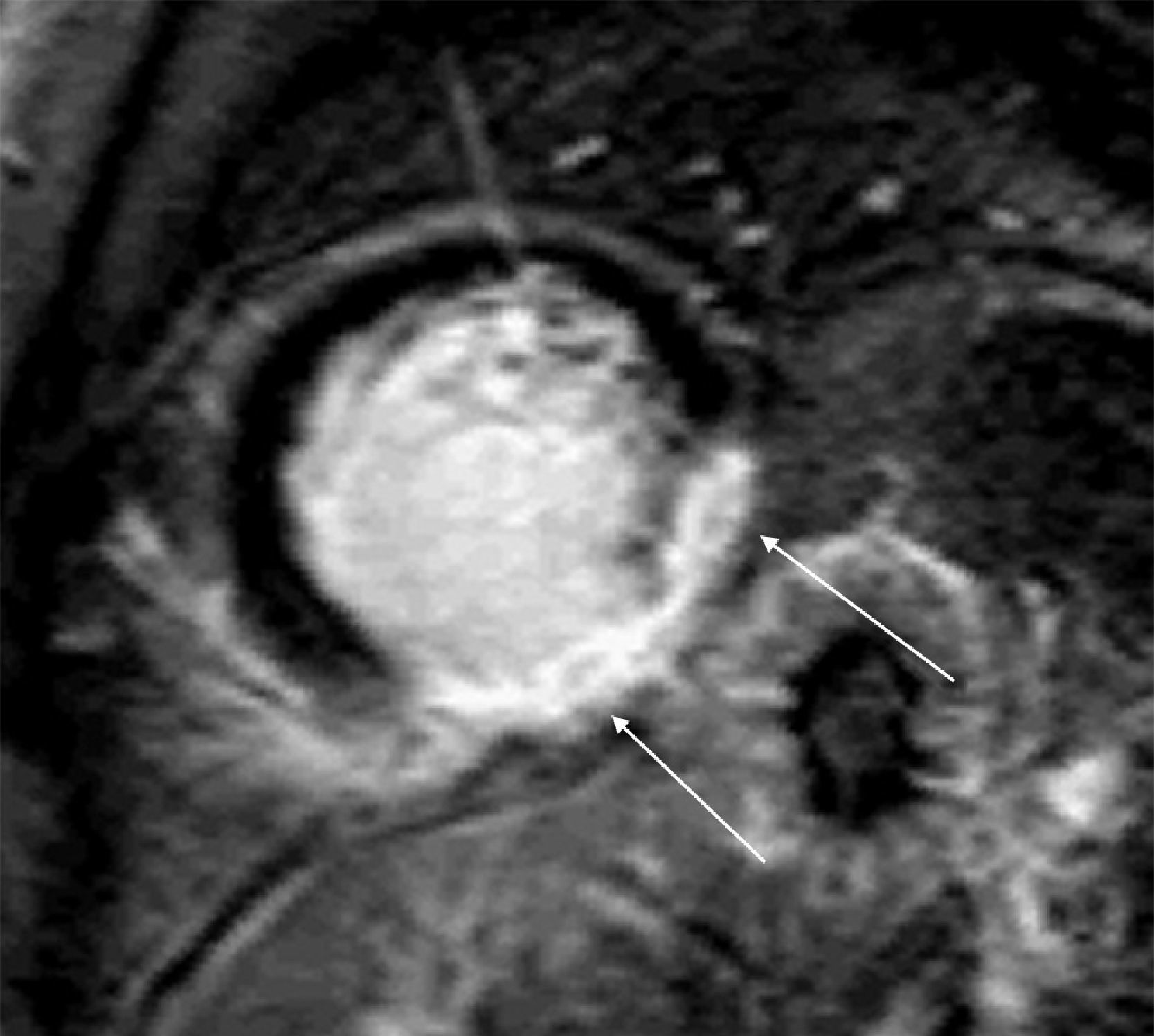
The patient was advised to maintain his commitment to quit smoking and was prescribed aspirin 100 mg daily, simvastatin 20 mg daily, perindopril 2 mg daily, nebivolol 2.5 mg daily and nitroglycerin 5 mg daily. He was referred for coronary angiography, which was performed on June 6, revealing irregularities of the right coronary artery (RCA) and what appeared to be a non-occlusive thrombus in its distal segment at the origin of the posterolateral artery (Figure 4) with preserved distal flow; there was no evidence of epicardial disease in the left anterior descending or circumflex arteries.
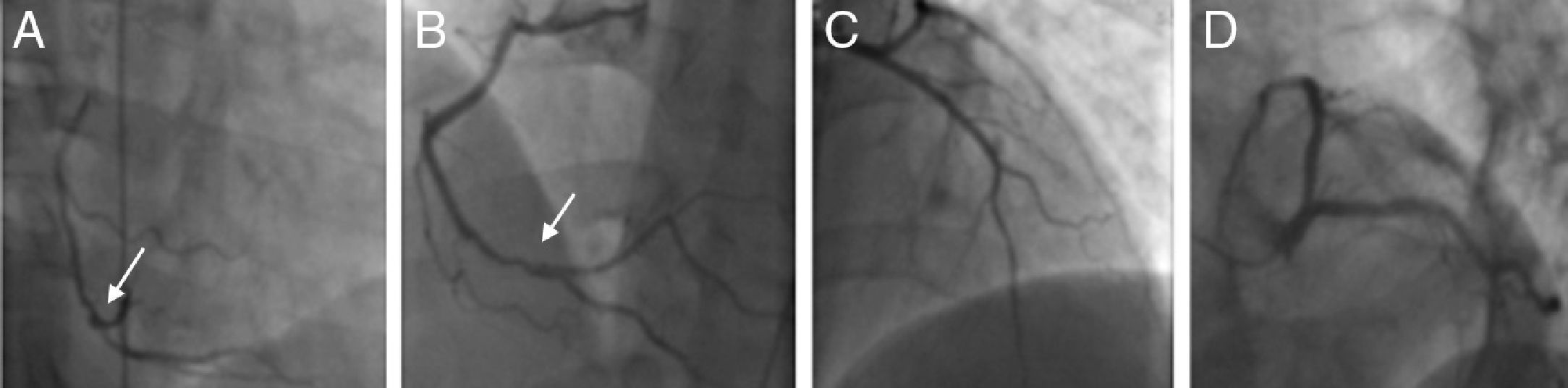
Coronary angiography on June 21, 2010. (A) and (B): Right coronary artery in right and left anterior oblique views, respectively, showing irregularities and what appears to be a non-occlusive thrombus in the distal segment (arrows); (C) and (D): left coronary artery in right anterior oblique view with cranial angulation and left anterior oblique (‘spider’) view, revealing no obstructive lesions.
His target INR was raised to 3–4; laboratory tests were repeated at this time and showed persistently high APA and positive ANA.
Autoimmune tests were also repeated six months later, which were strongly positive for APA and positive for ANA. Repeat brain MRI in December documented the evolution of the (presumed vascular) lesions, MRI angiography showing recanalization of the previously occluded branches of the RMCA.
The patient remained in New York Heart Association (NYHA) class I heart failure under optimized medical therapy (warfarin for a target INR of 3–4, simvastatin 20 mg daily, perindopril 2 mg daily, nebivolol 2.5 mg daily, furosemide 40 mg daily, and spironolactone 12.5 mg daily). TTE and TEE performed on January 1, 2011 showed a reduction in the size of the MV mass (0.33 cm×0.3 cm on TEE), but persistent moderate to severe LV systolic dysfunction (LVEF 33%). Repeat TTE on August 23 revealed similar LVEF to previous exams, but the MV mass was no longer visible.
In view of these findings, the patient was referred for an implantable cardioverter-defibrillator (ICD), implanted on September 23, without complications. During follow-up appropriate therapies were recorded for ventricular tachycardia in accordance with the device's programming, without causing symptoms.
Following ophthalmological evaluation, chronic hydroxychloroquine therapy was prescribed (400 mg daily) at an internal medicine consultation. Surveillance and correction of cardiovascular risk factors have been maintained, including smoking cessation and control of hypercholesterolemia with statins, as well as prophylaxis of bacterial endocarditis prior to procedures with a risk of bacteremia, and clinical, laboratory and echocardiographic monitoring of manifestations of SS, APS and SLE.
DiscussionSS is characterized by cerebral vascular lesions, most frequently in the territory of the middle cerebral artery,6–8 associated with LR, which usually precedes the onset of neurological symptoms, sometimes by decades, and intensifies during the acute phase of a neurological event,1 as seen in the case presented. LR results from reduced blood flow due to arterial occlusion and vasoconstriction of skin venules. It consists of an irregular purplish branching mesh-like or net-like skin discoloration, relatively unaffected by temperature, that generally begins on the trunk but may extend to contiguous areas and, rarely, to the extremities, which indicates extensive disease.1–3
In the case presented, SS was diagnosed in the context of a stroke, with subsequent investigation showing clinical and laboratory criteria for a diagnosis of APS (ischemic stroke and strongly positive APA in two measurements separated by more than 12 weeks). Patients with primary APS can also meet the criteria for SLE,2 and series have been published of patients meeting SLE criteria 3–10 years after being diagnosed with SS.5 In the present case, the patient fulfilled some criteria for a diagnosis of SLE, including thrombocytopenia (<100×103 platelets/μl) and positive ANA and APA (anticardiolipin and lupus anticoagulant antibodies). However, a diagnosis of SLE requires the presence of four criteria, which would be the case if photosensitivity or arthritis were included, but the patient's photosensitivity was intermittent and although he reported joint pain, arthritis was not confirmed. Since SS associated with APS would also explain thrombocytopenia and APA, it was decided to make a diagnosis of SS associated with APS and to direct treatment accordingly.
Two ischemic events occurred in the present case: stroke and myocardial infarction (MI), the latter under therapeutic levels of oral anticoagulation. Besides cerebral vascular lesions and LR, SS can also present with myocardial ischemia and fibrosis,1–6 which could explain both our patient's events. Recurrent ischemic events in different vascular territories associated with LV systolic dysfunction and wall motion abnormalities and RCA thrombus raise the possibility of a thrombotic or embolic etiology, due to the following mechanisms: (1) MI and stroke as thrombotic or embolic sequelae of APS and/or SS; (2) LV systolic dysfunction due to intracoronary thrombotic or embolic sequelae resulting from SS and/or APS, which could also be the source of cerebral embolism; or (3) the MV mass acting as a coronary and cerebral embolic source.
With regard to the stroke, the imaging findings of multiple bilateral small previous strokes, and involvement of the RMCA (a typical location), are suggestive of SS; this would be a long-standing etiology, but so would APS or the MV mass, since the RMCA occlusion is also typical of an embolic origin. Recanalization of the RMCA under anticoagulation as shown by MRI angiography suggests a thrombus rather than local fibrosis due to SS or embolization of the MV mass.
The description of an episode of intense angina (for which the patient did not seek medical assistance) followed by spontaneous remission in an anticoagulated patient, and the subsequent findings of transmural akinesia and fibrosis of the inferior and inferolateral walls, is clear evidence of a previous MI in the territory of the RCA, confirmed by coronary angiography, which revealed small atherosclerotic plaques and what appeared to be a thrombus in this artery. Possible causes of MI in this young patient include accelerated atherosclerosis due to APS or SLE in association with cardiovascular risk factors such as smoking and hypercholesterolemia, or embolization or local thrombosis in the context of APS, or embolization of the MV mass. Bearing in mind the clinical situation and angiographic findings, the most probable cause of the MI was local thrombotic or embolic events, which could be the result of SS or APS.
The angiographic findings were disproportionate to the severity of LV dysfunction, although this was present on the first TTE, on MRI (which documented fibrosis four months later), and on coronary angiography performed five months later when the patient was under anticoagulant therapy with therapeutic INR. The severity of LV dysfunction in the absence of epicardial disease in other coronary arteries, together with the failure to recover LV function with time, suggests that embolization or local thrombosis may have been more extensive in the past.
The MV mass observed could be a fibrous lesion of the type that has been identified in SS, particularly on the MV,1–5 or a non-infective vegetation, which has been reported in 6–10% of patients with APS and in 11% of those with SLE,14 when it is also known as Libman-Sacks endocarditis. Regression of such masses has been described with oral anticoagulation therapy, as has spontaneous regression.14 In the case presented, the mass may also have reduced in size due to embolization, which could have caused the cerebral vascular lesions or even the MI. The characteristics of the mass – sessile, of intermediate and heterogeneous echogenicity, with irregular, well-defined borders, and moving with the posterior MV leaflet – all suggest a non-infective vegetation, although the most common location in SLE is on the ventricular face of the MV,12 unlike in this case. The mass could also be a thrombus attached to the MV, which is a possible, although less frequent, presentation of APS,6 a hypothesis supported by its reduction under oral anticoagulation, its heterogeneous and intermediate echogenicity, and the hypointense T2-weighted signal. However, its well-defined borders and the absence of spontaneous contrast in the left chambers make this etiology less likely. Another possibility would be a benign tumor such as myxoma, which would be compatible with its appearance and echogenicity, but this would be an extremely unusual location.
Despite clinical evaluation in three different departments and a thorough diagnostic work-up, none of the above mechanisms could be completely confirmed.
In the assessment of the MV mass, MRI did not add significant information to that provided by TEE, which proved to be the most useful exam in this regard, but it did detect the presence of necrosis and thus enabled a diagnosis of previous MI following an episode of chest pain in a non-hospital setting and in a relatively young patient.
There is no effective treatment for SS. Anticoagulation is indicated in SS associated with APS, while antiplatelet therapy is the accepted treatment when not associated with APS,7 together with control of cardiovascular risk factors,1–5 which is equally important in patients with APS or SLE.13–16
In the case presented, anticoagulation with warfarin is the recommended initial therapy due to the risk of APS-related thrombosis or embolism of thrombi or the MV mass. Corticosteroid therapy is not indicated in the absence of consistent criteria for SLE or of renal damage, neurolupus, severe sustained cytopenia or pulmonary hemorrhage, and could aggravate the valve damage, prothrombotic state and LV dysfunction.2,16 Higher target INR (3–4) and hydroxychloroquine have been used for prophylaxis in APS and SLE patients with recurrent thrombotic events.14 The presence of >40 U/ml anticardiolipin antibodies, and SLE associated with these antibodies, are independent predictors of recurrence of thrombotic events at three years in patients with ASP,10 while positive anti-β2-glycoprotein-I antibodies at the time of the initial event shortens this period to less than 12 months.15 All these factors were present in our patient.
Surgical excision of the mass was not indicated in this case because it was small and was not causing significant valve dysfunction. However, prophylaxis of bacterial endocarditis prior to invasive procedures is recommended in patients with non-infective valve vegetations.15
In this case of a relatively young patient, already under anticoagulant therapy, diagnosed with SS and APS, in whom coronary angiography revealed no epicardial disease but documented a thrombus in the RCA with good distal flow and in whom MRI had shown non-viable myocardium in the territory of the RCA for which a thrombotic or embolic cause was most likely, the authors considered that ischemia or viability testing would not change the patient's treatment.
The referral for an ICD was for primary prevention of sudden death in a relatively young patient with ischemic heart disease, with transmural fibrosis documented by cardiac MRI and LVEF of <40% in assessments at 4, 12 and 20 months and in NYHA class I under optimized medical therapy. The appropriateness of this strategy is demonstrated by ICD therapy of episodes of ventricular tachycardia that could otherwise have been fatal.
ConclusionThe authors present an unusual case of SS associated with APS and possibly with SLE with two different forms of cardiac presentation: a valve mass and extensive MI in a relatively young man. The authors highlight the need to consider systemic disease such as APS, SLE or SS in cases of MI at young ages. The complexity of cardiovascular involvement in such diseases, as well as the difficulty in establishing the appropriate strategy, means an interdisciplinary approach is essential.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data and that all the patients included in the study received sufficient information and gave their written informed consent to participate in the study.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Privacy, informed consent, data protection and patient protectionThe patient provided written informed consent for the publication of this case report. The patient was not identified at any point in the study.
Conflicts of interestThe authors have no conflicts of interest to declare.
The authors thank the Radiology and Neuroradiology Department for their contribution to this case report and for providing the images.
Please cite this article as: Faustino A, Paiva L, Morgadinho A, et al. Síndroma antifosfolipídico associado a síndroma de Sneddon com envolvimento cardíaco: um diagnóstico desafiante. Rev Port Cardiol. 2014;33:115.e1–115.e7.
