
Anderson-Fabry disease is an X-linked lysosomal storage disorder caused by abnormalities of the GLA gene, which encodes the enzyme α-galactosidase A. A deficiency of this enzyme leads to the lysosomal accumulation of glycosphingolipids, which may cause left ventricular hypertrophy that is typically concentric and symmetric.
We present the case of a 60-year-old woman with symptoms of dyspnea, atypical chest pain and palpitations, in whom a transthoracic echocardiogram revealed an apical variant of hypertrophic cardiomyopathy. Analysis of specific sarcomeric genetic mutations was negative. The patient underwent a screening protocol for Anderson-Fabry disease, using a dried blood spot test, which was standard at our institution for patients with left ventricular hypertrophy. The enzymatic activity assay revealed reduced α-galactosidase A enzymatic activity. Molecular analysis identified a missense point mutation in the GLA gene (p.R118C).
This case report shows that Anderson-Fabry disease may cause an apical form of left ventricular hypertrophy. The diagnosis was only achieved because of systematic screening, which highlights the importance of screening for Anderson-Fabry disease in patients with unexplained left ventricular hypertrophy, including those presenting with more unusual patterns, such as apical variants of left ventricular hypertrophy. This case also supports the idea that the missense mutation R118C is indeed a true pathogenic mutation of Anderson-Fabry disease.
A doença de Anderson-Fabry é uma doença do lisossoma associada ao cromossoma X, causada por alterações no gene GLA que codifica a enzima α-galactosidase A. A deficiência desta enzima leva à acumulação de glicosfingolípidos, podendo causar hipertrofia ventricular esquerda, tipicamente concêntrica e simétrica.
Apresentamos o caso de uma mulher de 60 anos com queixas de dispneia, dor torácica atípica e palpitações, que realizou um ecocardiograma transtorácico que mostrou uma variante apical de miocardiopatia hipertrófica. A pesquisa de mutações nos genes das proteínas do sarcómero foi negativa. A doente foi incluída num protocolo de rastreio de doença de Anderson-Fabry, usando o teste da gota de sangue seca, para doentes com miocardiopatia hipertrófica. Foi documentada uma redução da atividade enzimática da enzima α-galactosidase A. A análise molecular identificou uma mutação missense do gene GLA (p.R118C).
Este caso clínico mostra que a doença de Anderson-Fabry se pode apresentar com um padrão de hipertrofia ventricular apical. Este diagnóstico apenas foi feito devido ao rastreio sistemático realizado em todos os doentes com hipertrofia ventricular esquerda de causa não esclarecida, incluindo mesmo os doentes com padrões de hipertrofia menos usuais. Este caso clínico também suporta a ideia de que a mutação missense R118C é de facto uma mutação patogénica de doença de Anderson-Fabry.
Anderson-Fabry disease (AFD) is an inborn error of metabolism caused by a genetic defect in the GLA gene, located in the Xq22 region of the X chromosome, which encodes the lysosomal enzyme α-galactosidase A.1 Absent or deficient activity of this enzyme leads to progressive lysosomal accumulation of neutral glycosphingolipids and α-galactosyl breakdown products in cells throughout the body.1 This has a profound effect on cellular function and leads to a wide range of manifestations in various organs, including the heart, kidneys and brain. Symptoms typically begin in childhood, although they become more frequent and severe with age, and patients may experience heart disease, renal failure or stroke.2 AFD is an X-linked disorder, and many years passed until it was recognized that heterozygous females were not merely carriers, but could indeed develop the full range of manifestations of the disease.3
Cardiac involvement is common (40–60%)4,5 and is currently the main cause of death in patients with AFD,6 which leads to structural and functional changes in the myocardium, conduction system and valves. The main cardiac manifestation is left ventricular (LV) hypertrophy.
It is currently recognized that the formerly reported annual incidence of AFD of 1 in 100 000 seriously underestimated the true prevalence of the disease.7 Recent prospective screening programs have revealed a much higher prevalence, of about 1 in 3100 hemizygous male newborns, mainly due to the diagnosis of late-onset phenotypes, such as late-onset hypertrophic cardiomyopathy.8,9 It has been estimated that AFD is responsible for at least 0.5% of cases of hypertrophic cardiomyopathy.10 LV hypertrophy secondary to AFD is typically concentric and symmetric.
We report an unusual case of an apical variant of LV hypertrophy in a patient with AFD carrying the missense mutation R118C, who was diagnosed during systematic screening of patients with unexplained LV hypertrophy.
Case reportA 60-year-old woman with a history of hypertension, dyslipidemia and chronic obstructive pulmonary disease presented to her general practitioner with dyspnea on moderate exertion (NYHA functional class II/IV), atypical chest pain and palpitations for several years. She underwent a transthoracic echocardiogram that revealed an apical variant of hypertrophic cardiomyopathy and she was referred to a cardiologist.
On clinical assessment the patient also complained of vertigo, hearing loss and tinnitus. There was no history of syncope or relevant family history. The physical examination was unremarkable.
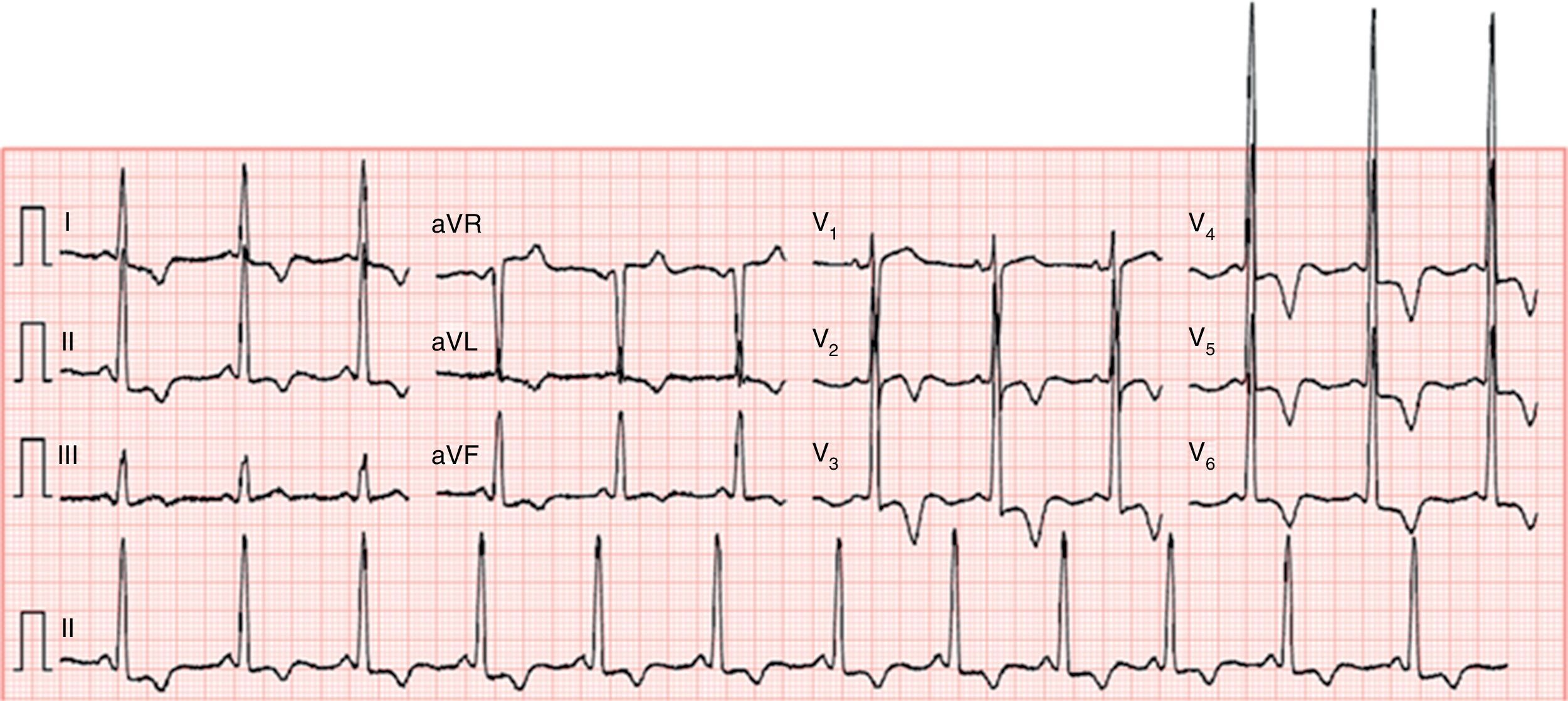
The electrocardiogram showed sinus rhythm (66 bpm), increased QRS voltage and diffuse, deep, symmetric T-wave inversion (Figure 1).
, increased QRS voltage and diffuse, deep, symmetric T-wave inversion.")
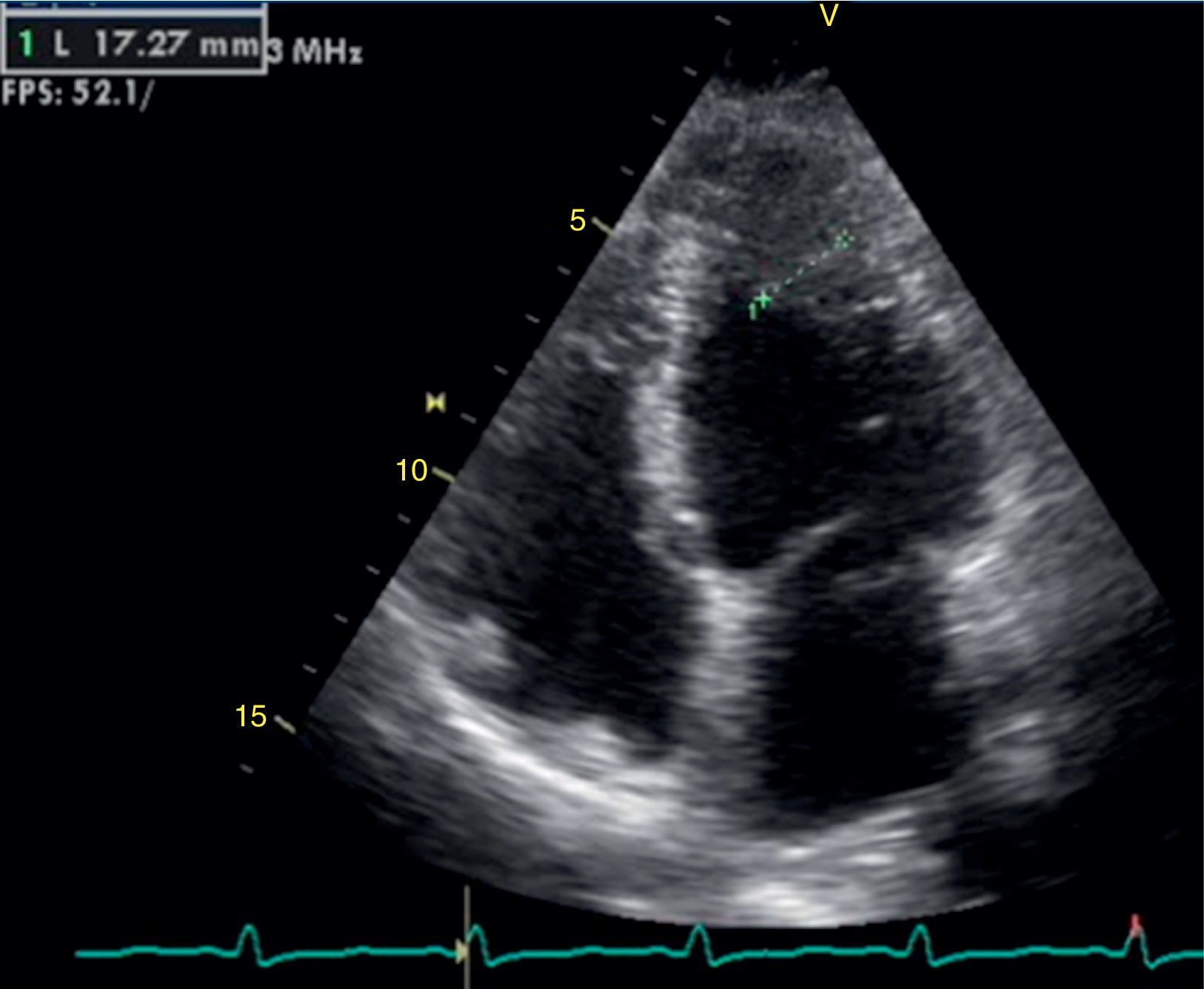
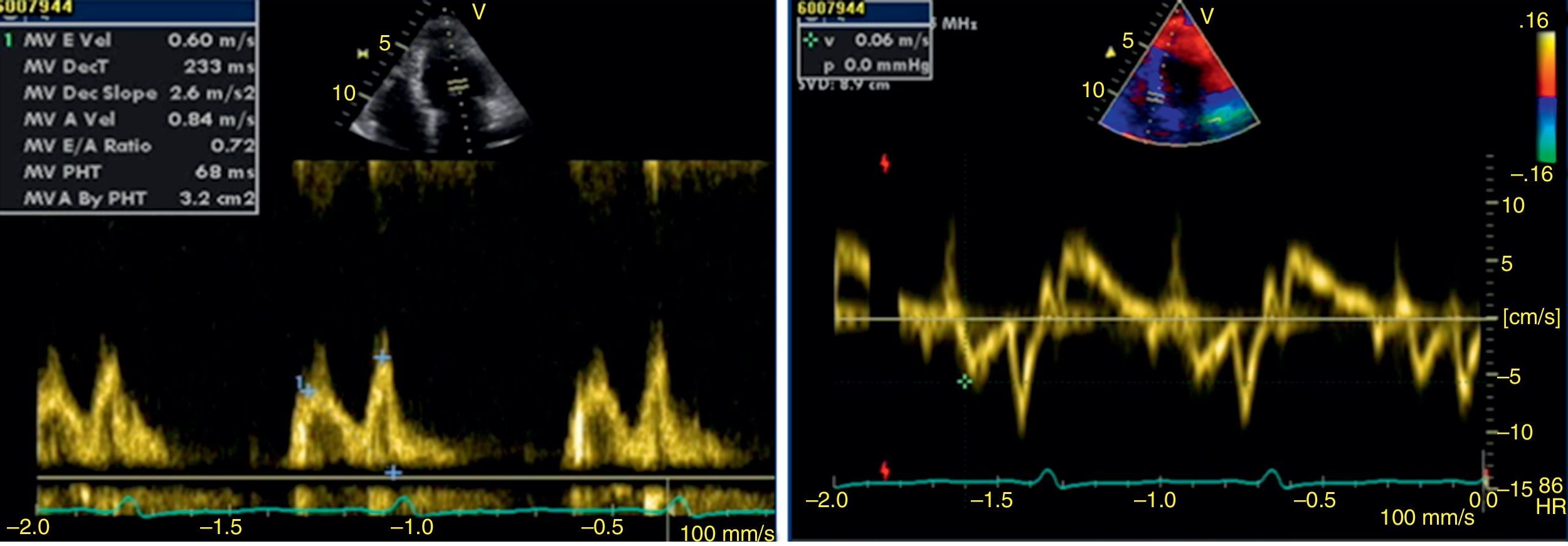
The transthoracic echocardiogram revealed apical LV hypertrophy with a typical spade-like geometry of the LV cavity at end-diastole (LV wall thickness 17 mm) (Figure 2) and LV diastolic dysfunction (impaired relaxation) (Figure 3) with normal ejection fraction. No structural or functional valve disease was present.

.")
There were no complex ventricular dysrhythmias on 24-hour Holter monitoring. A treadmill test was inconclusive for myocardial ischemia due to baseline electrocardiographic abnormalities, but there was a normal blood pressure response to exercise and no arrhythmias were found. Myocardial perfusion scintigraphy was normal.
The patient suffered from claustrophobia and refused to undergo cardiac magnetic resonance imaging.
A search for specific mutations in sarcomere protein genes was negative.
The patient was included in a screening protocol for AFD, using a dried blood spot test, which was standard at our institution for patients with unexplained LV hypertrophy. The enzymatic activity assay revealed reduced α-galactosidase A enzymatic activity (14 nmol/h/mg protein; normal range 36–80 nmol/h/mg protein). Molecular analysis revealed that the patient was heterozygous for a missense point mutation of the GLA gene (p.R118C).
A thorough multisystemic assessment was performed to search for other clinical features of AFD, but no renal, gastrointestinal, respiratory, cutaneous or ocular involvement was found.
Based on the finding of LV hypertrophy, the patient was referred for enzyme replacement therapy, but the decision of a national committee was not to start treatment but to keep the patient under clinical follow-up.
DiscussionTo our knowledge, this is one of the few case reports in the literature of an apical variant of LV hypertrophy in a female patient with AFD.11
The case shows that, although concentric and symmetric LV hypertrophy is the most typical pattern in AFD, other morphological patterns of LV hypertrophy may be found. It is estimated that asymmetric septal hypertrophy (more typically seen in hypertrophic cardiomyopathy caused by sarcomere protein gene mutations) accounts for 5% of cases.12
An apical form of LV hypertrophy should therefore not be an exclusion criterion for AFD screening. Indeed, this case was only diagnosed because it was included in a systematic screening protocol for AFD in patients with LV hypertrophy.
This case of an overt apical hypertrophic cardiomyopathy in a 60-year-old female also reinforces the current concept that female patients may develop as severe clinical manifestations as males, although usually later in life.
LV hypertrophy in AFD is usually associated with diastolic dysfunction, most commonly impaired LV relaxation, in the presence of preserved ejection fraction.13
As in this case, patients may present with a broad spectrum of cardiac signs and symptoms, including dyspnea, fatigue, chest pain, palpitations and syncope. Our patient had symptoms of dyspnea due to diastolic dysfunction. Arrhythmias, LV systolic dysfunction and valvular regurgitation may also contribute to heart failure. Anginal chest pain is explained by the imbalance caused by increased oxygen demand secondary to LV hypertrophy and impaired coronary supply due to functional hypoperfusion and/or fixed stenosis. Arrhythmias can explain the palpitations. Syncope can also occur secondary to tachyarrhythmias, particularly ventricular tachycardia, or bradyarrhythmias, including atrioventricular block.
This case also demonstrates the multisystemic nature of the condition and underlines the need for cardiologists today to be aware of the systemic manifestations of diseases affecting the heart in order to reach the correct diagnosis.
Like our patient, most patients with AFD experience hearing loss at some time during their lives (85% of males over 50 years of age and 75% of females over 60). The majority suffer from neurosensorial hearing loss, severe enough to require the use of hearing aids.14
Nevertheless, our patient did not show respiratory tract involvement (reduction in forced expiratory volume in one second or in carbon monoxide diffusion capacity, or increase in mean residual volume), peripheral nervous system manifestations (such as neuropathic pain or hypohydrosis), central nervous system manifestations (ischemic stroke or epilepsy – although due to claustrophobia brain magnetic resonance imaging was not performed to document asymptomatic white matter lesions), renal impairment (microalbuminuria, decreased glomerular filtration rate), gastrointestinal complaints (diarrhea, nausea, vomiting, abdominal bloating), or cutaneous (angiokeratomas) or ocular involvement (cornea verticillata or cataracts).
AFD can be diagnosed in males by demonstrating deficiency of α-galactosidase A activity. In heterozygous females, α-galactosidase A enzymatic activity may be within the normal range, requiring molecular analysis for a definite diagnosis.
Although this patient was female, reduced plasma α-galactosidase A enzymatic activity was found. The diagnosis of AFD was then established by molecular analysis, which identified a missense point mutation in the GLA gene (p.R118C). This mutation has been described in patients with AFD.8,15–18 A high prevalence of this mutation was found in Portugal and Spain in one study, in which the possibility of a polymorphism was excluded because this mutation was not found in 240 alleles from healthy controls. A founder effect was also excluded as the mutation was spread across Spain.17 This mutation is associated with relatively high residual α-galactosidase A enzymatic activity.8,17 However, our patient had reduced enzymatic activity, in spite of being female, which also supports the idea that this mutation is a true pathogenic mutation; it has also been found in patients presenting angiokeratomas,15 stroke16 and renal disease under hemodialysis.17
As far as we know, this is the first report of LV hypertrophy in a patient with AFD carrying the missense mutation R118C. In this case, LV hypertrophy was apical. The other case of AFD associated with an apical form of LV hypertrophy occurred in a patient carrying the mutation R227Q.11
Enzyme replacement therapy with agalsidase is indicated in patients with LV hypertrophy secondary to AFD because it has been shown to reduce LV wall thickness and mass and to improve LV function.18
ConclusionThis case shows that AFD may cause an apical form of left ventricular hypertrophy. The diagnosis was only achieved because of systematic screening, which highlights the importance of screening for Anderson-Fabry disease in patients with unexplained LV hypertrophy, including those presenting with more unusual patterns, such as apical variants of LV hypertrophy.
As far as we know, this is the first report of LV hypertrophy in a patient with AFD carrying the missense mutation R118C, which supports the idea that this is a true pathogenic mutation, despite the relatively high residual enzymatic activity frequently encountered in individuals with this mutation.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data and that all the patients included in the study received sufficient information and gave their written informed consent to participate in the study.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.
