
A Origem Anómala da Artéria Coronária Esquerda da Artéria Pulmonar é uma cardiopatia congénita rara e uma causa de isquemia miocárdica em idade pediátrica. A maioria dos casos não diagnosticados morre no primeiro ano de vida, sendo necessária a formação de uma extensa rede de colaterais para permitir a sobrevivência. O diagnóstico não é linear exigindo elevada suspeição clínica. Os autores apresentam o caso de uma criança de oito anos de idade, de raça negra, assintomática, referenciada de Cabo Verde para esclarecimento de dilatação e disfunção do ventrículo esquerdo com fluxos turbulentos sistodiastólicos a nível do septo interventricular. Dos antecedentes destacava‐se quadro de insuficiência cardíaca diagnosticada aos três meses de idade, com avaliação posterior compatível com miocardiopatia dilatada. Teve melhoria clínica após início de terapêutica anticongestiva que mantinha na altura da admissão no nosso Centro. Ecocardiograficamente suspeitou‐se de Origem Anómala da Artéria Coronária Esquerda da Artéria Pulmonar, sendo este diagnóstico confirmado através de angiotomografia computorizada e cateterismo cardíaco. A doente foi submetida, com sucesso, a implantação direta da artéria coronária esquerda na aorta permitindo criar um sistema de perfusão coronário duplo. Este caso ilustra uma forma incomum de uma patologia rara que sobreviveu sem diagnóstico após o primeiro ano de vida. Reforça igualmente a importância da multimodalidade de imagem nestes casos.
Anomalous Origin of the Left Coronary Artery from the Pulmonary Artery is a rare congenital heart disease and a cause of myocardial ischemia during childhood. Most undiagnosed cases die in the first year of life as an extensive collateral network is essential for survival. The diagnosis requires a high index of clinical suspicion. The authors present the case of an 8‐year‐old black asymptomatic child referred from Cape Verde Island in order to clarify left ventricular dilatation and dysfunction with systo‐diastolic turbulent flows observed at the interventricular septum. At the age of 3 months, she was diagnosed with heart failure, in the context of showing dilated cardiomyopathy. She was managed and clinically improved with anticongestive therapy, which she was still taking at the time of admission to our Center. The echocardiogram findings suggested Anomalous Origin of the Left Coronary Artery from the Pulmonary Artery and the diagnosis was confirmed by computerized angiotomography and cardiac catheterization. The patient was successfully submitted to direct implantation of the left coronary artery into the aorta, allowing the creation of a double coronary perfusion system. This case illustrates an unusual presentation of a rare pathology that survived without a diagnosis after the first year of life. It also reinforces the importance of multimodality image screening in these cases.
A Origem Anómala da Artéria Coronária Esquerda (ACE) da Artéria Pulmonar (Anomalous Origin of the Left Coronary Artery from the Pulmonary Artery ‐ ALCAPA), corresponde a 0,24 – 0,46% das cardiopatias congénitas1. Apesar de rara, é a anomalia coronária congénita mais frequente. Na ausência de tratamento, apenas 10‐15% dos casos atinge a idade adulta, dependendo esta sobrevivência do desenvolvimento de circulação colateral inter‐coronária2.
O ecocardiograma é o método diagnóstico de primeira linha, podendo, contudo, ser necessário recorrer a outros exames imagiológicos para definição superior da anatomia coronária. A angiotomografia coronária computorizada define a origem das artérias coronárias e a relação destas com as estruturas adjacentes. Permite, assim, efetuar medições indispensáveis para o cirurgião planear a abordagem cirúrgica. A coronariografia seletiva mantém‐se, para muitos autores o método gold‐standard para o planeamento cirúrgico.
A cirurgia é o tratamento de eleição nos pacientes com ALCAPA, podendo tornar‐se emergente nas crianças muito sintomáticas. O tratamento ideal consiste na criação de um sistema de perfusão coronário duplo. No tipo infantil, quando a anatomia é favorável, opta‐se pela correção mais anatómica que consiste na implantação direta da ACE na aorta. Uma alternativa é a criação de um conduto intrapulmonar do óstio coronário esquerdo até à aorta (procedimento de Takeuchi). Este último apresenta como complicações tardias estenose pulmonar, fuga intra‐pulmonar, estenose e regurgitação aórtica3.
No adulto, geralmente realiza‐se revascularização miocárdica da ACE e laqueação da sua origem na artéria pulmonar. A vantagem desta técnica é evitar a manipulação da raiz da aorta, mas tal deve ser contrabalançado com o eventual risco de uma menor durabilidade do enxerto.
Após correção cirúrgica a evolução é geralmente favorável.
Caso ClínicoOs autores apresentam o caso de uma criança do sexo feminino, raça negra, de 8 anos de idade, evacuada de Cabo Verde para esclarecimento de cardiopatia.
Iniciou clínica de insuficiência cardíaca aos três meses, com diagnóstico ecocardiográfico aos 11 meses de miocardiopatia dilatada, pelo que iniciou terapêutica anticongestiva com melhoria do quadro clínico.
Foi evacuada aos oito anos para o nosso Centro para melhor esclarecimento diagnóstico e orientação terapêutica. Encontrava‐se assintomática, apresentando à auscultação um sopro contínuo no bordo para‐esternal superior esquerdo.
A telerradiografia torácica apresentava ligeira cardiomegalia e o eletrocardiograma tinha critérios de hipertrofia ventricular esquerda com sinais de isquemia subendocárdica da parede lateral e septal, sem evidência de necrose.
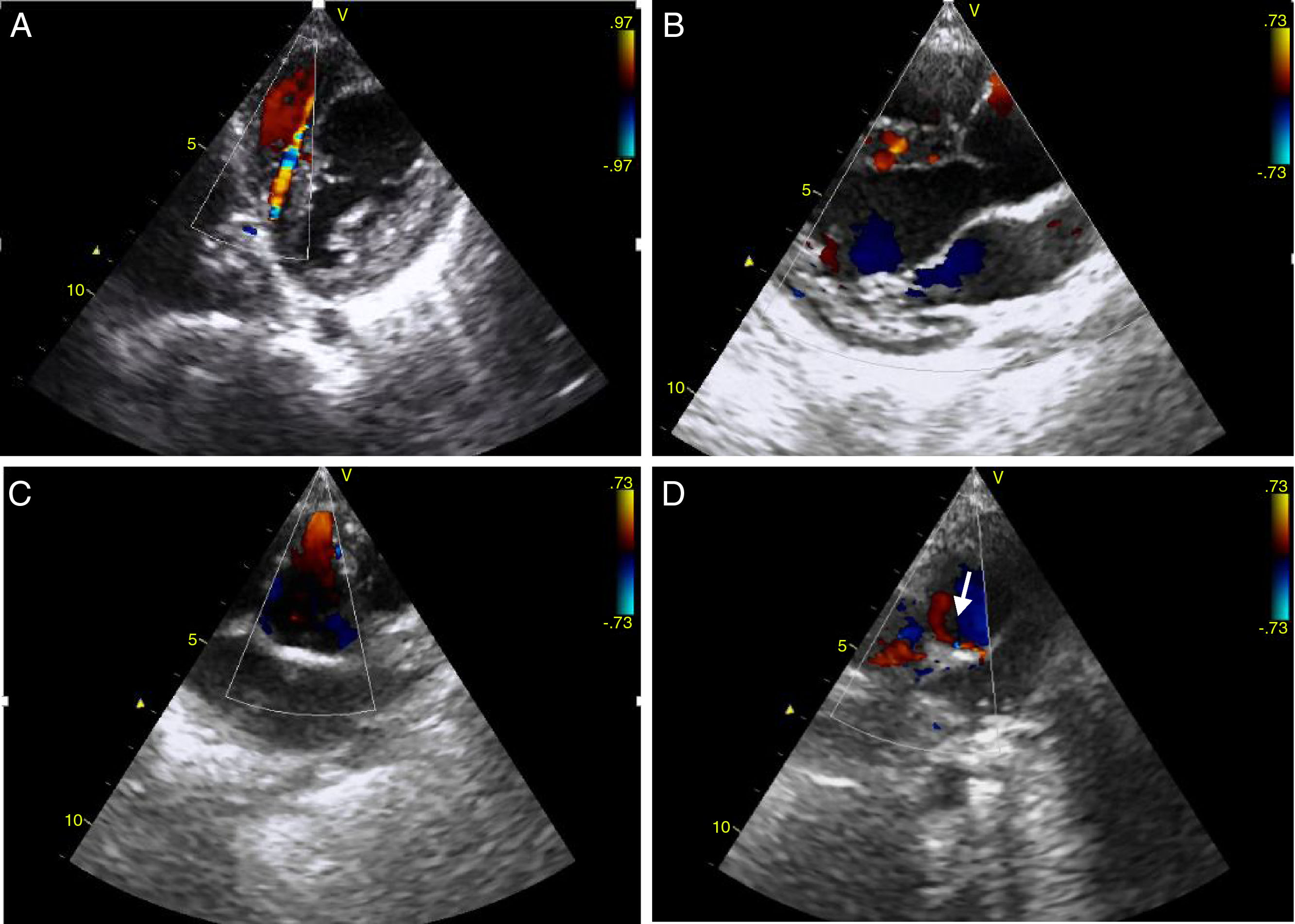
Ecocardiograficamente observou‐se ventrículo esquerdo dilatado com região apical hipocinética e aneurismática; ecogenicidade aumentada do aparelho subvalvular mitral compatível com fibroelastose secundária a isquemia do músculo papilar. O estudo Doppler identificou múltiplos fluxos sistodiastólicos interventriculares com velocidade máxima < 1 m/s (Figura 1A e 1B). Não apresentava regurgitação valvular mitral e a fração de ejeção calculada por método de Simpson biplano foi de 48%. A artéria coronária direita (ACD) encontrava‐se ectasiada (Figura 1C), não sendo possível determinar a origem da ACE. Em incidência paraesternal eixo curto, foi visível fluxo retrógrado no tronco da artéria pulmonar (TAP), logo após a válvula pulmonar, sugestivo de runoff da ACE (Figura 1D, seta).
 e (B). Em paraesternal eixo curto de vasos evidencia‐se fluxo anterógrado na artéria coronária direita que apresenta dilatação da sua porção proximal (C) e fluxo retrógrado no tronco da artéria pulmonar (seta) correspondente ao run‐off da artéria coronária esquerda (D).")
Ecocardiograma transtorácico com Doppler codificado a cor
Em incidência paraesternal eixo curto e paraesternal eixo longo são visíveis fluxos sistodiastólicos a nível interventricular correspondendo a extensa rede de colaterais (A) e (B).
Em paraesternal eixo curto de vasos evidencia‐se fluxo anterógrado na artéria coronária direita que apresenta dilatação da sua porção proximal (C) e fluxo retrógrado no tronco da artéria pulmonar (seta) correspondente ao run‐off da artéria coronária esquerda (D).
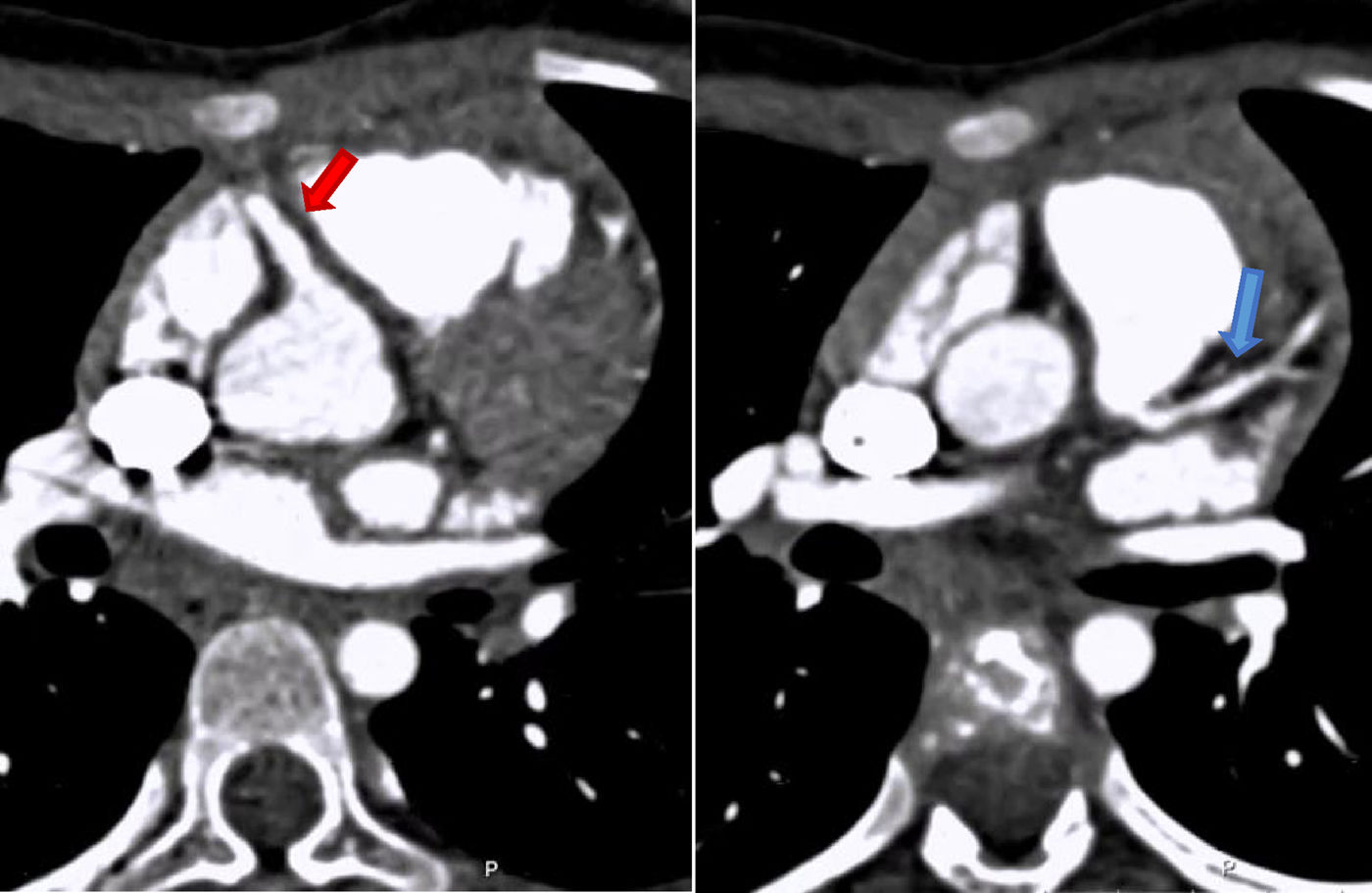
Para confirmação diagnóstica realizou Angio – TAC coronária que mostrou tronco comum da ACE com origem na vertente inferior esquerda do TAP, a cerca de 10mm da raiz da aorta (Figuras 2 e 3). Esta distância levantou dúvidas sobre a possibilidade de transferência direta da ACE para aorta. Por apresentar períodos de frequências cardíacas mais elevadas (apesar de fármaco cronotrópico negativo) e artefactos de imagem secundários a movimento, apenas foi possível definir o trajeto proximal das coronárias.
 e a esquerda com origem na face póstero‐lateral esquerda do tronco da artéria pulmonar (seta azul).")
 e circunflexa (não dominante) (B). Estes vasos têm permeabilidade preservada na porção visualizada – sua metade proximal.")
Coronariografia por TC multidetectores ‐ 64 cortes, ECG gated. Reformatações multiplanares e curvas
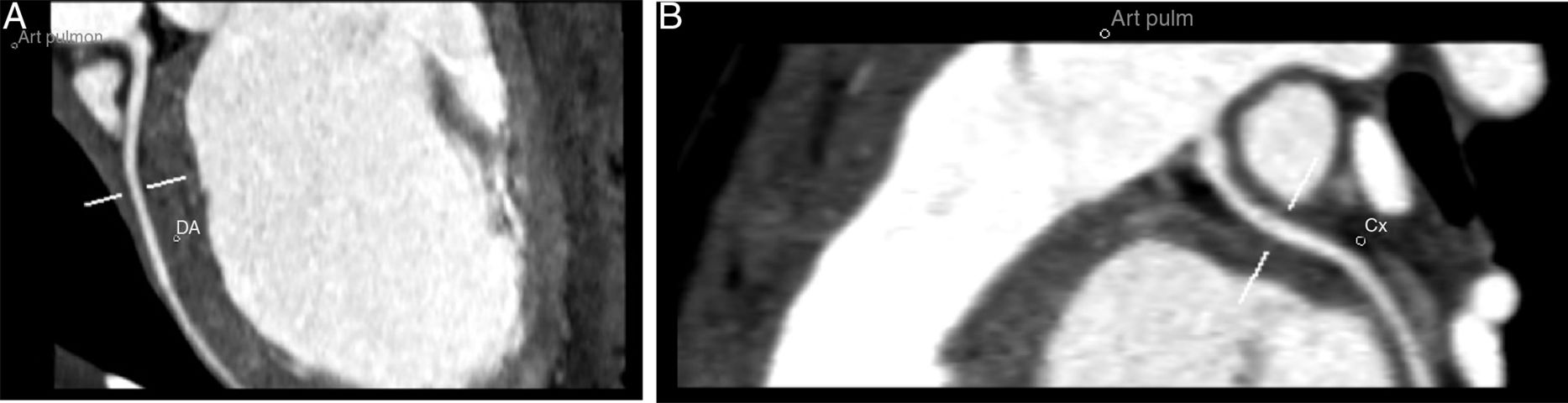
Tronco comum da artéria coronária esquerda com origem na vertente inferior esquerda do tronco da artéria pulmonar. O tronco comum apresenta um calibre de 2,6mm e uma extensão de cerca de 3mm, tem contornos regulares e permeabilidade preservada.
O tronco comum apresenta a normal bifurcação em artéria descendente anterior (A) e circunflexa (não dominante) (B). Estes vasos têm permeabilidade preservada na porção visualizada – sua metade proximal.
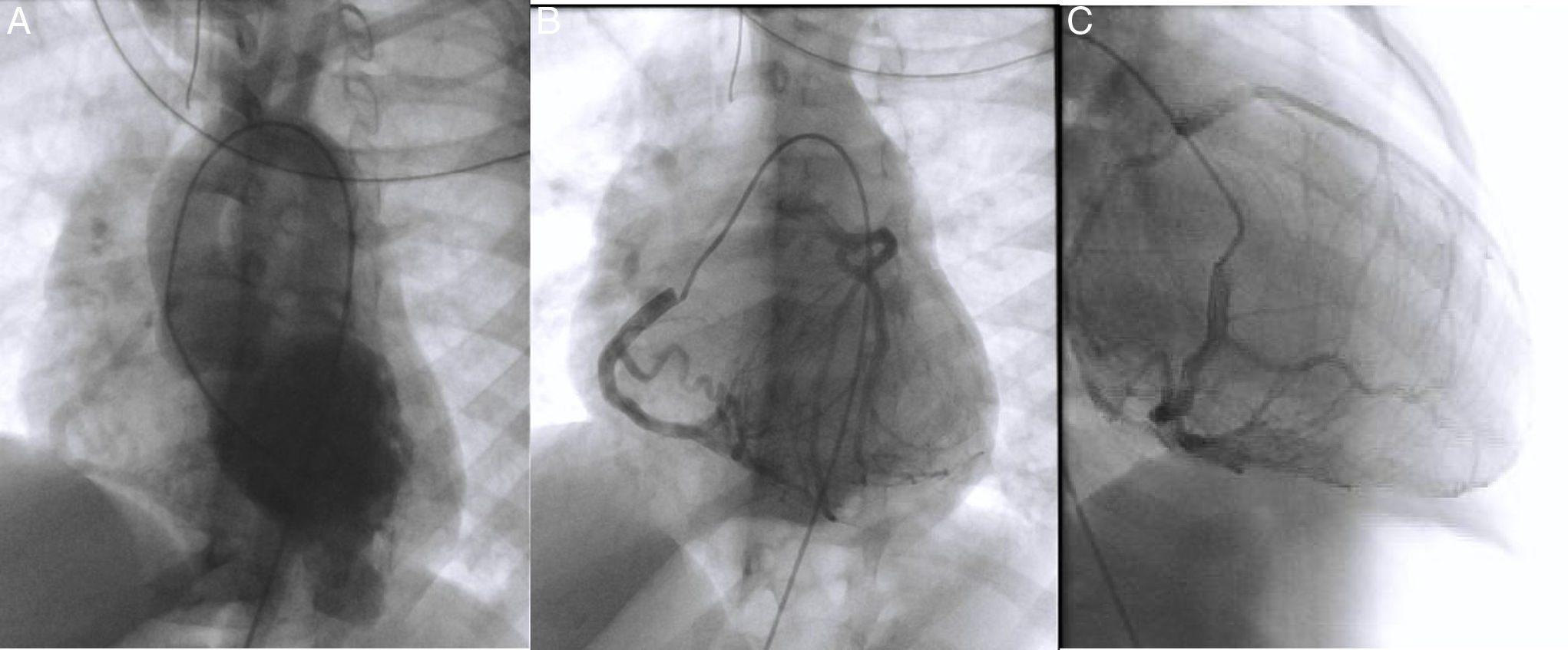
Realizou, também, cateterismo cardíaco que confirmou a origem anómala da ACE a partir do TAP, assim como perfusão retrógrada da ACE através da ACD por uma extensa rede de vasos colaterais e posterior drenagem no TAP. Detetou‐se ainda redução luminal da porção distal da artéria descendente anterior (Figura 4B e 4C). A ventriculografia esquerda evidenciou dilatação aneurismática da região apical do VE (Figura 4A) e disfunção sistólica ligeira. Verificou‐se que a pressão telediastólica do VE estava elevada com pressões e resistência vascular pulmonar normais. Tentou‐se cateterização seletiva da ACE a partir do TAP sem sucesso e uma angiografia no TAP não evidenciou opacificação da ACE.
. Coronariografia direita em projeção póstero anterior e oblíqua anterior direita mostra coronária dilatada que comunica e preenche a artéria coronária esquerda através de rede colateral extensa. O tronco da artéria coronária esquerda origina‐se do tronco da artéria pulmonar. O fluxo de sangue faz‐se: aorta‐> artéria coronária direita‐> colaterais‐> artéria coronária esquerda‐> artéria pulmonar. A porção distal da descendente anterior encontra‐se mal preenchida (B) e (C).")
Cateterismo cardíaco diagnóstico.
Ventriculografia esquerda em projeção póstero anterior revela região apical do ventrículo aneurismática, preenchimento da aorta e da artéria coronária direita (A).
Coronariografia direita em projeção póstero anterior e oblíqua anterior direita mostra coronária dilatada que comunica e preenche a artéria coronária esquerda através de rede colateral extensa. O tronco da artéria coronária esquerda origina‐se do tronco da artéria pulmonar. O fluxo de sangue faz‐se: aorta‐> artéria coronária direita‐> colaterais‐> artéria coronária esquerda‐> artéria pulmonar. A porção distal da descendente anterior encontra‐se mal preenchida (B) e (C).
Após discussão com o cirurgião, foi submetida a reimplantação direta do tronco comum da ACE na raiz da aorta, com boa evolução pós cirúrgica.
A criança manteve as alterações eletrocardiográficas que demonstrava à admissão. Para excluir défices de perfusão após a reimplantação da coronária realizou, antes de regressar ao país de origem (cerca de um mês após a cirurgia), uma prova de esforço segundo protocolo de Bruce que excluiu disritmias ou agravamento das alterações de repolarização da parede septal e lateral com o exercício. A avaliação ecocardiográfica mostrou fluxo adequado na coronária reimplantada, normalização das dimensões das cavidades esquerdas, com disfunção sistólica do VE mais ligeira.
DiscussãoDe acordo com a expressão clínica da doença, podemos classificar a ALCAPA em dois tipos: o infantil e o adulto. Na vida fetal, as pressões pulmonares elevadas possibilitam um fluxo anterógrado na ACE, permitindo uma boa adaptação in utero. Após o nascimento, a diminuição gradual das pressões e resistências vasculares pulmonares resulta numa pressão de perfusão coronária progressivamente mais baixa, culminando na inversão do fluxo na ACE.
No tipo infantil, o mais comum, após a reversão do fluxo na ACE não se forma uma rede colateral entre as coronárias, que permanecem de diâmetro normal. A diminuição do fornecimento de sangue ao miocárdio alimentado pelo território da ACE traduz‐se em cardiomiopatia isquémica que cursa com 90% de mortalidade no primeiro ano de vida.
No tipo adulto, o fluxo reverso na ACE estimula a formação de uma circulação colateral inter‐coronária com dilatação gradual das coronárias, possibilitando uma pressão de enchimento adequada da ACE a partir da ACD. Progressivamente, a dilatação das coronárias origina fluxo de sangue preferencialmente para a artéria pulmonar em detrimento do sistema de elevada resistência do miocárdio, ocorrendo o fenómeno de roubo coronário. Neste subgrupo, desenvolve‐se doença miocárdica isquémica crónica potenciando o risco de morte súbita por disritmias.
Realça‐se que a função ventricular depende mais da pressão de perfusão das coronárias do que da saturação de oxigénio do sangue que transportam. Por isso, contrariamente ao ALCAPA, na transposição simples das grandes artérias, que se caracteriza por baixos índices de saturação de oxigénio nas artérias coronárias mas pressão de perfusão coronária normal, a função ventricular esquerda é preservada.
O nosso caso é o de uma criança com ALCAPA que sobreviveu devido ao desenvolvimento de uma extensa circulação colateral coronariana direita‐esquerda. O início dos sintomas aos três meses poderá ter coincidido com a diminuição progressiva da resistência vascular pulmonar e o encerramento do canal arterial. Na avaliação ecocardiográfica, no país de origem, foi diagnosticada miocardiopatia dilatada de causa não esclarecida. Trata‐se, de facto, de um diagnóstico que necessita de um elevado índice de suspeição e de exames complementares diferenciados para a sua confirmação.
A melhoria da disfunção ventricular deveu‐se provavelmente, à rede colateral extensa que se desenvolveu durante o período mais crítico, permitindo manter uma pressão de perfusão coronária e aporte de oxigénio ao miocárdio em risco.
À admissão no nosso Centro, esta criança apresentava‐se assintomática, refletindo a adaptação favorável proporcionada pela revascularização natural. O sopro contínuo auscultado está relacionado possivelmente com o runoff da ACE para o TAP. Ao ecocardiograma apresentava as alterações típicas do subtipo adulto da ALCAPA: fluxos sistodiastólicos intraventriculares (rede colateral extensa que alimentava a ACE); dilatação e disfunção ventricular esquerda (secundária a isquemia do miocárdio) e não origem da ACE na aorta. No entanto, está descrito que poderá ocorrer uma falsa imagem da ACE a emergir da aorta. Nestes casos a direção anómala (retrógrada) do fluxo a nível da ACE ajuda ao diagnóstico correto.
A tomografia coronária apresenta limitações técnicas em idade pediátrica, pelo que pode ser necessário complementar o estudo com angiografia coronária conforme ocorreu neste caso.
No nosso cateterismo caracterizámos o fluxo de sangue coronário e toda a anatomia coronária. Verificámos redução luminal da porção distal da artéria descendente anterior com consequente insuficiente perfusão do seu território distal com região aneurismática e hipocinesia do ápex.
À semelhança do que foi a decisão da equipa de cirurgia do nosso Centro, a correção deverá favorecer a criação de um sistema de perfusão coronário duplo.
Após esta intervenção, é expectável que a função e dilatação ventricular esquerda melhorem gradualmente. No caso descrito, apesar do pouco tempo de seguimento pós‐operatório, já se observou normalização das dimensões do VE e melhoria da disfunção ventricular sistólica.
ConclusãoA ALCAPA é uma cardiopatia rara associada a alta mortalidade. A história natural dos poucos casos que sobrevivem após o primeiro ano de vida sem diagnóstico inclui isquemia crónica, disfunção ventricular e disritmias potencialmente fatais. A multimodalidade de exames complementares permite uma melhor acuidade diagnóstica e fornece dados fundamentais ao cirurgião. A correção cirúrgica deve ser considerada em todos os pacientes, preferindo‐se uma abordagem que crie um sistema coronário duplo. O prognóstico após a cirurgia é geralmente favorável.
Conflito de interessesOs autores declaram não haver conflito de interesses.
Os autores agradecem ao Dr. Rui Catarino pela colaboração na execução e escolha das imagens do presente trabalho.
